
Abstract
Multiple sclerosis is a disease characterized by inflammation and demyelination located in the central nervous system. Experimental autoimmune encephalomyelitis (EAE) is the most common animal model for multiple sclerosis (MS). Although the roles of T cells in MS/EAE have been well investigated, little is known about the functions of other immune cells in the neuroinflammation model. Here we found that an essential cytokine transforming growth factor β (TGF-β) which could mediate the differentiation of Th17/regulatory T cells was implicated in the natural killer (NK) cells’ activity in EAE. In EAE mice, TGF-β expression was first increased at the onset and then decreased at the peak, but the expressions of TGF-β receptors and downstream molecules were not affected in EAE. When we immunized the mice with MOG antigen, it was revealed that TGF-β treatment reduced susceptibility to EAE with a lower clinical score than the control mice without TGF-β. Consistently, inflammatory cytokine production was reduced in the TGF-β treated group, especially with downregulated pathogenic interleukin-17 in the central nervous system tissue. Furthermore, TGF-β could increase the transcription level of NK cell marker NCR1 both in the spleen and in the CNS without changing other T cell markers. Meanwhile TGF-β promoted the proliferation of NK cell proliferation. Taken together, our data demonstrated that TGF-β could confer protection against EAE model in mice through NK cells, which would be useful for the clinical therapy of MS.
Introduction
Multiple sclerosis (MS) is a disease in which inflammation in the central nervous system (CNS) results in the destruction of myelin sheath 1,2 . Although inflammation combined with environmental and genetic factors plays an essential role in the pathogenesis of MS, the precise mechanisms of MS remain unclear. The breakdown of the blood–brain barrier is the onset symptom of MS, then the inflammatory cells and cytokines invade the CNS to destroy myelin, and the remyelination process determines the recovery after each exacerbation 3 . Acute relapses respond well to corticosteroid therapy, providing further evidence that inflammation is central to the disease process 4 . Experimental autoimmune encephalomyelitis (EAE) is a mouse or rat T cell-mediated autoimmune disease model in the CNS used to simulate the human MS condition 5 . The transforming growth factor β (TGF-β) family of growth factors controls the homeostasis and development in multiple organs. The TGF-β signal transduction network involving kinases and their substrates, the SMAD proteins, has been partly elucidated in the past few years 6,7 . After receiving the signaling from TGF-β via the receptors, SMAD can move into the nucleus to activate target gene transcription in association with DNA-binding partners such as transcriptional factors 8 . Therefore, either loss or specific mutations in these pathways could cause various forms of human immune disorders and cancer. It is also reported that TGF-β could regulate regulatory T (Treg) cells’ function to inhibit autoimmune diseases mediated by Th17 9 . For the cell-specific mechanism, distinct cells display multifunctional characters in human MS and murine EAE. Although CD4 T cells play a central role in this process, other immune cells, including B cells 10 and natural killer (NK) cells 11 , also contribute to the development and determine the severity of the disease. When mice were deprived of NK cells by antibody treatment before immunization, they developed a more serious form of EAE associated with relapse. This phenotype was supported by augmentation of T cell proliferation and production of Th1 cytokines in response to MOG antigen 12 . However, less is known about the mechanisms underlying NK cells in the control of MS/EAE. NK cells distinguish between normal healthy cells and abnormal cells by using a sophisticated repertoire of cell surface receptors that control their activation or inhibitory functions 13 . These receptors on NK cells, including rodent Ly49 receptors, human killer cell immunoglobulin-like receptors, and conserved CD94/NKG2 receptor family, could specifically recognize MHC class I molecules or related ligands or host encoded non-MHC ligands 14 .
Materials and Methods
Reagent
Purified TGF-β was purchased from R&D Systems China Co (Wuhan, China). The dose of TGF-β protein for in vivo administration was 200 μg with intraperitoneal (i.p.) injection and for in vitro treatment was 10 ng/ml.
Ethical Approval
The animal care and the experiments described here were carried out in agreement with the guidelines set by the Institutional Animal Investigation Committee of Animal Biosafety Level 3 Laboratory of Wuhan University (Wuhan, China). The protocol was approved by the Committee on the Ethics of Animal Experiments of Wuhan University. Animals were housed under a 12-h light/dark cycle, and were kept in the same animal care facility for the duration of the study. All efforts were made to minimize any suffering and to reduce the total number of animals used.
EAE Induction
To induce EAE in C57BL/6 mice, female mice were immunized subcutaneously in the flanks with 50 μg MOG35–55(MEVGWYRSPFSRVVHLYRNGK) in complete freund’s adjuvant (CFA) containing 200 μg of Mycobacterium tuberculosis H37RA (Difco, Detroit, MI, USA), followed by i.p. injection of 100 ng of pertussis toxin (List Biochemicals, Campbell, CA, USA) 15 . Clinical assessment of EAE was performed according to the following criteria from a previous study 16 .
Reverse Transcription-Quantitative Polymerase Chain Reaction
Total RNA was extracted from the cells using the RNeasy mini kit (QIAGEN China Co., Shanghai, China), followed by complementary DNA synthesis using the Superscript III first strand synthesis kit (Invitrogen; Thermo Fisher Scientific, Waltham, MA, USA) at 25°C for 10 min, 50°C for 30 min, and 85°C for 5 min. Quantitative polymerase chain reaction was performed on a Bio-Rad amplifier using the Bio-Rad real time polymerase chain reaction (PCR) mix, including SYBR Green dye (both Bio-Rad, Hercules, CA, USA). The following thermocycling conditions were used for the PCR: 50°C for 2 min, 10 min at 95°C; 40 cycles of 95°C for 15 s and 60°C for 1 min. Data were analyzed using the Cq value normalized to the endogenous reference gene GAPDH 17 .
Enzyme-linked immunosorbent assay
Mouse enzyme-linked immunosorbent assay (ELISA) kits for cytokine detection in the sera or homogenate were obtained from R&D Systems China Co. To detect low levels of cytokines in the samples, a standard curve was obtained by diluted standard reagents.
MTT Assay
Cells were incubated with 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium (MTT) reagent (5 µg/ml final concentration) at 37°C for 4 h. Formazan was solubilized by adding 100 µl dimethyl sulfoxide into each well. The extent of formazan production was determined by an ELISA reader at a wavelength of 550 nm, while 630 nm served as the reference wavelength. The results were calculated according to the manufacturer’s instructions (Vybrant™ MTT Cell Proliferation Assay Kit, Thermo Fisher Scientific).
Results
EAE Model TGF-β Expression was Declined Without Affecting its Receptors and Downstream
Consistent with the clinical MS data 18 , we found significant changes of TGF-β 1 expression with an increased level in early EAE but reduced level in the later stage, in both spinal cord and brain (Fig. 1A). However, the other molecules associated with the TGF-β RI and TGF-β RII (Fig. 1B) in spinal cord; meanwhile, the downstream SMAD subtype expression was not influenced in the EAE model (Fig. 1C).

TGF-β expression in experimental autoimmune encephalomyelitis (EAE). (A) The expression of TGF-β on different days during EAE. (B) The expression of TGF-β receptors on different days during EAE. (C) The expression of TGF-β downstream SMAD molecules on day 14 during EAE.
TGF-β Treatment Attenuated EAE
Next we tested the function of TGF-β in the EAE model and detected that TGF-β treated mice were resistant to neuroinflammation in EAE with reduced clinical score (Fig. 2A) in the i.p. injection. During a 15-day observation, both the TGF-β cytokine treated mice and the control group without TGF-β intervention displayed the symptoms of EAE after model establishment from day 8, but TGF-β treatment in vivo could attenuate disease severity, displayed by lower clinical score, suggesting TGF-β suppresses EAE development with certain mechanisms. Considering MS/EAE is a type of autoimmune disease with increased inflammatory cytokine production, we next detected the cytokine profiles in the EAE mice in the presence of TGF-β. Consistent with the phenotype demonstrated by clinical scores, TGF-β inhibited inflammation of EAE with decreased serum levels of interleukin (IL)-2/IL-6/IL-17 (Fig. 2B). Furthermore, as Th17 cells and its cytokine IL-17 play central roles in the EAE model, we observed decreased IL-17 production in the CNS homogenate (spinal cord and brain) from EAE mice with TGF-β treatment, suggesting TGF-β specifically prevents neuroinflammation in the disease development (Fig. 2C).

The effect of TGF-β on the experimental autoimmune encephalomyelitis (EAE) model. (A) Clinical score of EAE model following treatment with TGF-β. EAE clinical score shown on Y axis means: 0, no clinical signs; 1, partially limp tail; 2, paralyzed tail; 3, hind limb paresis, uncoordinated movement; 4, one hind limb paralyzed; 5, both hind limbs paralyzed; 6, hind limbs paralyzed, weakness in forelimbs; 7, hind limbs paralyzed, one forelimb paralyzed; 8, hind limbs paralyzed, both forelimbs paralyzed; 9, moribund; 10, death. (B) Serum cytokine production in each group was measured by enzyme-linked immunosorbent assay (ELISA). (C) CNS IL-17 production in each group was measured by ELISA.
The NK Marker NCR1 Was Increased by the TGF-β In Vivo
When we treated the mice with high dose TGF-β recombinant protein, we found that the mouse NK cell marker NCR1 was increased not only in spleen and splenocyte (Fig. 3A and B), but also in the CNS tissue (spinal cord in Fig. 3C and brain in Fig. 3D). However, TGF-β failed to alter the T cell markers’ expression in CNS such as CD3, CD4 and CD8 (Fig. 3C), suggesting the specific cytokine could selectively promote the proliferation and infiltration of NK cells without affecting infiltrated T cells in the CNS of EAE mice.
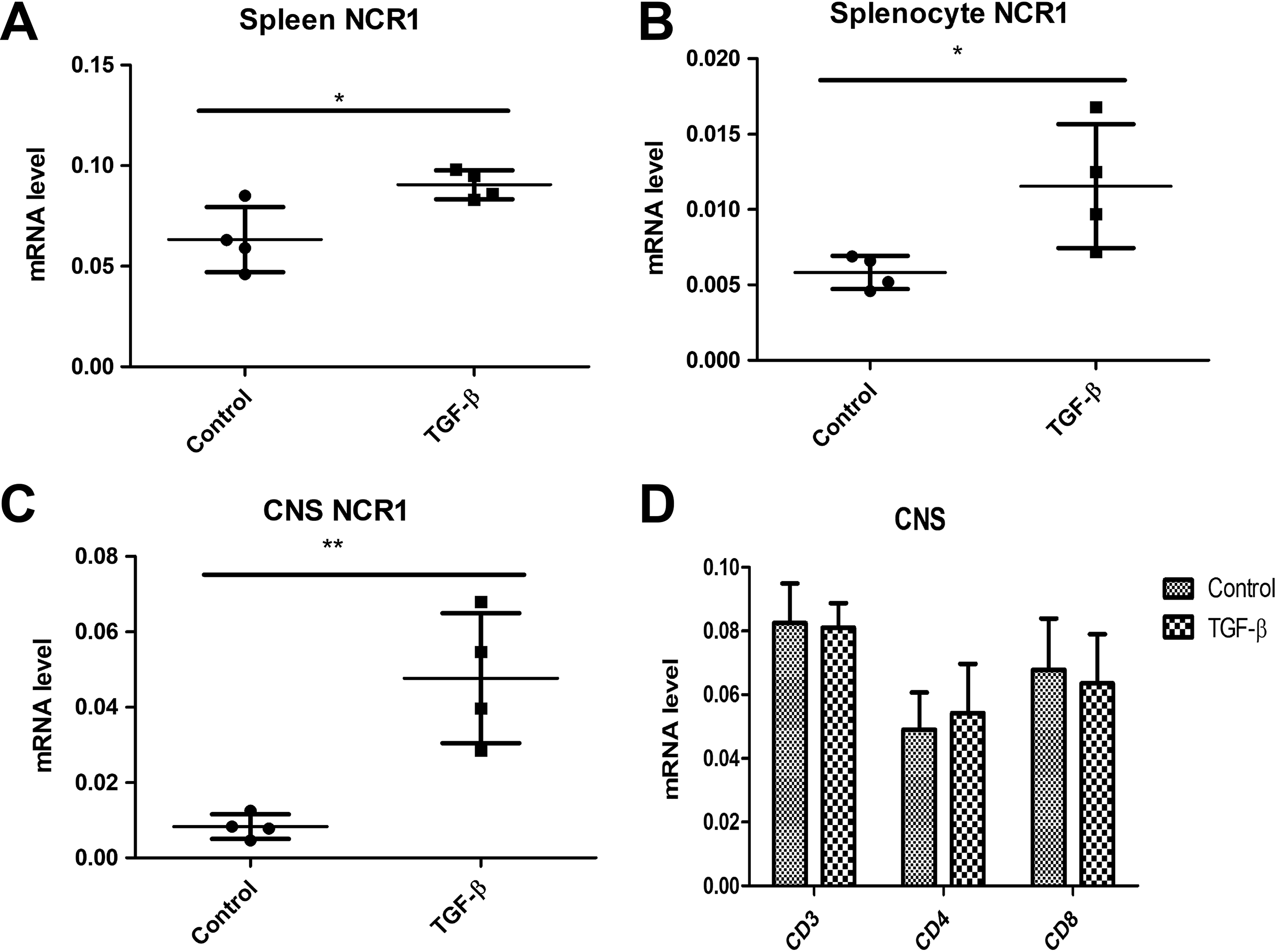
TGF-β regulated natural killer (NK) cell marker NCR1 transcription in vivo. (A) Spleen NCR1 mRNA expression was determined by quantitative polymerase chain reaction (qPCR). (B) Splenocyte NCR1 mRNA expression was determined by qPCR. (C) CNS NCR1 mRNA expression was determined by qPCR. (D) CNS CD molecules’ mRNA expression was determined by qPCR.
TGF-β Increases the NK Cell Proliferation In Vitro
To further confirm the effect of TGF-β in vitro, we evaluated the proliferation of NK cells after TGF-β pretreatment. We found that TGF-β enhanced NK cell proliferation in a dose dependent manner (Fig. 4A). Taken together, our data demonstrated that TGF-β confers protection on the murine EAE model through specific NK cell activity.

TGF-β regulated NK cell proliferation in vitro. (A) MTT assay of proliferation of NK cells isolated from the control mice treated with TGF-β in vitro. (B) A model of the biological function of TGF-β in the experimental autoimmune encephalomyelitis targeting NK cells.
Discussion
Th17 and anti-inflammatory Treg cells often keep a balance in the homeostasis of a healthy state, while in the EAE environment, the proinflammatory cytokine IL-17 produced by Th17 cells driven by IL-23 could induce the tissue lesions in the CNS, whereas the IL-17 deficient mice were not sensitive to the EAE model 19 –21 . In these two major subsets of CD4 T cells, TGF-β played an important role in the in vitro differentiation: TGF-β plus IL-6 as the factors responsible for differentiation of Th17 cells (IL-6 is added to suppress FOXP3 expression but upregulates the Th17 related RORA and RORC genes). However, in the absence of IL-6, Treg cells can be induced by the TGF-β alone in vitro 22 . Signalings through TGF-β and its receptors, including TGF-β RI/TGF-β RII are essential for both Th17 and Treg development 23,24 . However, the roles of TGF-β and NK cells remain uncertain in the EAE model. In most clinical MS patients, increased pro-inflammatory cytokines were combined with decreased production of TGF-β, which plays an important role as anti-inflammatory cytokine in the manifestation of MS. Defective production of anti-inflammatory cytokine TGF-β by T cell lines was detected in patients with active MS 25 . In this current study we also found that TGF-β confers protection against the mouse model of MS. TGF-β was downregulated in the peak of the disease, and the administration of TGF-β protein can rescue the severity of EAE in a dose dependent manner.
Next, we wondered whether TGF ameliorates EAE through T cell signaling, in the TGF-SMAD signaling; the downstream transcriptional factors could be activated by the TGF through the receptors for gene transcription. However, no T cell markers’ (CD3, CD4,CD8) mRNA expression was affected in the CNS region by TGF treatment, suggesting that TGF may regulate a unique pathway in other immune cells. Considering the reported data that NCR1 in NK cells was regulated in the MS white matter lesions by regulating innate immunity 26 , we attempted to clarify that NCR1 and NK cells are functionally activated in the EAE model associated with the TGF-β signaling. Natural cytotoxicity receptors (NCRs) are activating receptors expressed on the NK cells. The human NCR family includes NKp30, NKp44, and NKp46, but mice express only the homologue protein NKp46, named NCR1 27 . In fact, pathogenic or regulatory roles of NK cells with the cell marker NCR1 are implicated in many diseases, for example, it is reported that NCR1 plays an essential role in type 1 diabetes. Targeting NK cells’ reactivity by employing NCR1 antibody could lower the incidence of diabetes in both the non-obese diabetes model and the low-dose streptozotocin induced diabetes model 28 . In another study, TGF-β was found to be associated with NK cell activity in cancer. In the murine model of head and neck cancer, TGF-β could downregulate the NKG2D, which is the NK-activating receptor 29 . In our study we first evaluated the phenotype of TGF-β in the EAE model and then measured the effect of TGF-β on the NK cells’ activating receptor NCR1. In vitro data also supported that TGF-β promoted NK cell proliferation, and we speculate that TGF-β might trigger NCR1 expression as well as NK cell proliferation through the TGF-R-SMAD pathway (Fig. 4B). At the transcriptional level, NCR1/NKp46 expression could be directly regulated by the runt-related transcription factor 3 (RUNX3) in vitro 30 , while the TGF-β regulating NCR1 in NK through the RUNX3 still needs further study (Fig. 4B). Taken together, our findings could provide an important therapeutic approach based on immune regulation for the murine EAE model and clinical MS.
Footnotes
Ethical Approval
The study was approved by the Institutional Animal Investigation Committee of Animal Biosafety Level 3 Laboratory of Wuhan University (Wuhan, China).
Statement of Human and Animal Rights
All of the experimental procedures involving animals were conducted in accordance with the Institutional Animal Investigation Committee of Animal Biosafety Level 3 Laboratory of Wuhan University (Wuhan, China).
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.