
Abstract
There is growing evidence that stem and progenitor cells exert regenerative actions by means of paracrine factors. In line with these notions, we recently demonstrated that endothelial progenitor cell (EPC)-derived conditioned medium (EPC-CM) substantially increased viability of brain microvascular cells. In the present study, we aimed at investigating whether EPC-CM supports cell survival of cultured striatal progenitor cells. For that purpose, primary cultures from fetal rat embryonic (E14) ganglionic eminence were prepared and grown for 7 days in vitro (DIV). EPC-CM was administered from DIV5–7. Treatment of the striatal cultures with EPC-CM resulted in significantly increased densities of GABA-immunoreactive (-ir) neurons. Inhibition of mitogen-activated protein kinase and phosphatidylinositol-3-kinase, but not of the ROCK pathway, significantly attenuated the EPC-CM induced increase in GABA-ir cell densities. Similar results were observed when EPC-CM was subjected to proteolytic digestion and lipid extraction. Furthermore, inhibition of translation abolished the EPC-CM induced effects. Importantly, EPC-CM displayed neuroprotection against 3-nitropropionic acid induced toxicity. These findings demonstrate that EPC-derived paracrine factors substantially promote survival and/or differentiation of cultured striatal progenitor cells involving both proteinaceous factors and lipidic factors. In sum, EPC-CM constituents might lead to a novel cell-free therapeutic strategy to challenge neuronal degeneration.
Keywords
Introduction
Neurodegenerative diseases pose a growing burden and, despite much effort, effective interventions are currently still lacking. Progresses in stem cell research have raised the prospect that nervous tissue might be regenerated by exploiting the characteristic proliferative and differentiation properties of these cells. Stem cells are generally considered as a tool for replacing injured cells, but increasing attention has recently been given to the capacity of stem and progenitor cells to secrete a wide array of factors. Indeed, it is the humoral actions, rather than the transdifferentiation/engraftment, of stem cells that are now believed to be the main actors in the scenario of tissue regeneration. In this regard, there is compelling evidence that paracrine factors released by bone marrow-derived endothelial precursors support the viability of different tissues 1,2 . We have previously shown that the endothelial progenitor cell (EPC) “secretome” in the form of conditioned medium (EPC-CM) has high tissue regenerative potential 3 –5 . EPCs and their soluble factors have also been used successfully in traumatic brain injury, ischemic stroke, and white matter damage models, but these effects have usually been interpreted in the context of neurovascular repair 2,6 –8 . Moreover, transplantation of neuronal progenitors from ganglionic eminence (GE) for the treatment of Huntington’s disease (HD) has shown encouraging results, and great efforts are currently being exerted to improve transplantation protocols in animal models of the disease 9 . In the present work, we aimed to investigate the effect of EPC-CM more specifically on differentiating primary striatal progenitor cells from rat fetuses. In particular, we analyzed whether EPC-CM might influence the yield and survival of GABAergic neurons, and attempted to characterize the factors and pathways involved in the activity of EPC-CM.
Materials and Methods
Animals
Time pregnant Wistar rats were purchased from Janvier Labs (Le Genest-Saint-Isle, France) and housed in a 12-hour light-dark cycle with food and water ad libitum. All experiments were carried out in the light phase and in accordance with the guidelines of the Animal Research Ethics Committee of Canton Bern, Switzerland, and the University of Bern Animal Care and Use Committee, Switzerland (study No. 6/12 and 10/15).
Preparation and Culture of Rat Dissociated Fetal GE Cultures
GE cultures were prepared from embryonic day 14 (E14) rat fetuses as described previously
10
. In brief, pregnant Wistar rats were anesthetized using gas inhalation (4.5–5% isoflurane, 75% N2O, 20% O2; Peachtree Corners, GA, USA) followed by an i.p. injection of a mix of ketamine (120 mg/kg; Vetoquinol AG, Ittigen, Switzerland) and xylaxine (20 mg/kg; Vetoquinol AG). The fetuses were separated by caesarean section and the dams decapitated and exsanguinated immediately thereafter. The fetuses were then euthanized by decapitation and the GE dissected out of the brain using a stereoscopic microscope. The GE explants were dissociated mechanically and plated at a density of 0.5 embryos/well (seeding density of 600 viable cells per mm2) into 24-well plates with a glass insert (glass coverslips of 12 mm diameter, Assistent, Sandheim, Germany) pre-coated with poly-
EPC Culture and CM Preparations
EPC were isolated from peripheral blood mononuclear cells from healthy human anonymous donors as previously described 11 . The buffy coats employed for this procedure were purchased from the Interregional Transfusion Centre of the Swiss Red Cross (Bern, Switzerland), and no experimental approval or informed consent was needed. Moreover, all samples were handled according to the regulations and guidelines of the University Hospital Bern. Mononuclear cells were prepared by centrifugation through a density gradient on Lymphoprep (Axis-Shield, Dundee, UK) and plated on fibronectin (Sigma, Buchs, Switzerland)-coated 6-well plastic dishes (Falcon) at a density of 1 million cells/cm2 using endothelial cell basal medium-2 (EBM-2) (Lonza, Basel, Switzerland) supplemented with endothelial growth medium BulletKit and 5% FBS (Lonza, Basel, Switzerland). After 4 days in culture, adherent cells were passaged at a density of 0.1 million cells/cm2 and cultured through day 7 to obtain early outgrowth EPC. To produce EPC-CM, EPC were incubated for 48 hours under hypoxic conditions (1.5% O2, 5% CO2, 93.5% N2) using a humidified gas-sorted anoxic incubator-gloved box (InVivo2 400, Ruskinn Technologies, Bridgend, UK) in growth factor-free EBM-2 containing 1% FBS since we have previously observed that these culture conditions enhance soluble factor secretion by EPC 3 . After incubation, EPC-CM was collected and centrifuged at 1000 rpm at 4°C, the supernatant was sterile filtered and snap-frozen until use 3 . The EBM-2 containing 1% fetal bovine serum (FBS) was treated in parallel to the cell cultures and served as control medium (unconditioned medium). Throughout all the experiments, EPC-CM and control medium were processed simultaneously with the same treatments.
Conditioned Medium Treatments
To eliminate the proteinaceous content in the CM, the CM was treated with proteinase K as described earlier 5,12 . In brief, aliquots of EBM-2 or CM medium were incubated with agarose beads-proteinase K (Sigma, Buchs, Switzerland; 5 U) in 15 ml polypropylene test tubes (BD Falcon, Allschwil, Switzerland) for 4 h at 37°C in a rotating roller drum. Thereafter, the tubes were centrifuged 5 min at 2000 rpm to pellet the agarose beads-proteinase K, and the proteinase K-free supernatant was carefully collected and used for subsequent experiments. The treatment resulted in degradation of proteins as assessed in Coomassie Blue (Sigma, Buchs, Switzerland)-stained gels 5,12 (Fig. 1).

Representative SDS-PAGE of untreated and proteinase K-treated (prot-K) control (Ctrl) and EPC-CM media stained with Coomassie Blue. Note that protein signals including BSA (arrowhead) are significantly reduced in both media after 4 h of proteinase K-treatment.
Control medium and EPC-CM were treated with chloroform as previously described 5,13 to remove the lipidic components. In brief, EBM-2 or CM medium was mixed with equal volumes of chloroform. Thereafter, the medium was centrifuged for 5 min at 3000 rpm, the aqueous phase was collected and the procedure was repeated. The medium was then transferred into an open 1 cm Petri dish and the traces of chloroform were left to evaporate in the sterile hood for 10 min. Finally, the volume of water lost due to the evaporation was reconstituted with distilled sterile water.
Treatments
Culture wells were assigned randomly to the different treatment groups and incubated from DIV5 until the end of the experimental period at DIV7. To investigate the effects of CM on GE cultures, CM was added to the cultures and compared to control cultures treated with EBM-2. To further investigate the potential signaling involved in the effects observed in the first experiments, the specific phosphoinositide-3-kinase/activated kinase (PI3K/AKT) inhibitor LY294002 (Gibco, Reinach, Switzerland) (10 µM), the specific mitogen-activated protein kinase kinase/extracellular regulated kinase (MEK/ERK) inhibitor PD98059 (Gibco, Reinach, Switzerland) (10 µM) and the selective Rho-associated coiled-coil containing kinases (ROCK) inhibitor Y27632 (Sigma, Buchs, Switzerland) (10 µM) were added 30 min prior the treatment with EBM-2 and EPC-CM, respectively. To investigate whether translation might interfere with EPC-CM mediated effects on GABA-ergic cell density cycloheximide (Sigma, Buchs, Switzerland) (10 µM) was added to the culture medium at DIV5 with or without concurrent EPC-CM addition. Moreover, based on our results of EPC-CM mediated effects on microvascular endothelial cells 5 , we investigated whether the observed effects after EPC-CM treatment are attributed to the lipidic or the proteinaceous content of the EPC-CM. For that purpose, cultures were treated with chloroform-extracted EPC-CM and proteinase-K-treated EPC-CM, EBM-2 was exposed to the same treatments and served as controls.
The effects on neuroprotection were assessed by the treatment of the cultures with 3-nitropropionic acid (3-NP; Sigma, Buchs, Switzerland; 5 mM) from DIV5-7 in combination with either EBM-2 or EPC-CM. In addition, we tested whether exposure of cultures to EPC-CM from DIV2-5 prior to the insult altered the effects during the 3-NP treatment. 3-NP is an irreversible inhibitor of mitochondrial respiratory complex II succinate dehydrogenase, resulting in energy depletion through disruption of the electron transport chain and eventually in the death of GABA-ergic neurons that closely mimics HD neuropathology 14 –16 .
Cell Viability Assay
Cell viability was assessed as previously described with minor modifications 4 . Briefly, cultures were treated as described above. At DIV7, the experimental media were replaced with incubation medium and the number of viable cells was assessed by the Presto Blue assay (Invitrogen, Reinach, Switzerland) using a microplate reader (Ex. 560 nm, Em. 590 nm; VarioSkan, ThermoFisher Scientific, Reinach, Switzerland).
Western Blotting and Protein Measurements
The cells were lysed in RIPA buffer (ThermoFisher Scientific, Reinach, Switzerland) containing protease inhibitor cocktail set V (Sigma, Buchs, Switzerland), 1 mM phenylmethylsulfonyl fluoride (PMSF, Sigma, Buchs, Switzerland) 1 µg/ml leupeptin (Sigma, Buchs, Switzerland) and 1 mM sodium orthovanadate (Sigma, Buchs, Switzerland). The protein concentration was determined in two aliquots per lysate using the Pierce BCA Protein Assay Kit (ThermoFisher Scientific, Reinach, Switzerland). Total protein content in control cultures was 355.8 ± 7.1 µg per culture. The Western blotting was carried out according to the method of Laemmli 17 with some modifications. Equal amounts of the proteins were run on a 12% polyacrylamide gel and transferred to a PVDF membrane (Bio-Rad Laboratories AG, Cressier, Switzerland). The transfer efficiency was checked by Coomassie Blue staining of the gels. Blots were blocked for 45 min in 5% fat-free milk powder in TBS-Tween 0.01% and incubated with the same solution overnight with primary antibodies (mouse anti-glial fibrillary acidic protein (GFAP), 1:500, Millipore, Zug, Switzerland; mouse anti-proliferating cell nuclear antigen (PCNA), 1:1000, Sigma, Buchs, Switzerland; rabbit anti-α-tubulin, 1:2000, Sigma, Buchs, Switzerland). After washing in TBS-Tween 0.01% the blots were incubated for 1 h with antibody-peroxidase conjugates (anti-mouse or anti-rabbit, 1:10,000; Jackson ImmunoResearch, Ely, UK) in 5% fat-free milk powder in TBS-Tween 0.01%. Subsequent to washing the blots were visualized with the enhanced chemiluminescence substrate kit (ThermoFisher Scientific, Reinach, Switzerland) using the Fusion Pulse TS System (Vilber Lourmat, Collégien, France). Bands were quantified on 8bit converted images using the Fiji software and normalized against α-tubulin. Eight cultures from four independent experiments were included for the analyses.
Immunocytochemistry
To visualize GABAergic neurons, the cell cultures were fixed for 30 min in 4% paraformaldehyde, washed and blocked with 10% horse serum in 0.1% Triton-X-100/PBS. Thereafter, the cells were incubated overnight with the primary antibody rabbit anti-GABA (1:5000; Sigma, Buchs, Switzerland) in 0.1% Triton-X-100/PBS with 2.5% horse serum at 4°C. Subsequent to washes in PBS, the cultures were incubated for 2 h with biotinylated anti-rabbit IgG (1:200, Vector Laboratories, Servion, Switzerland). Endogenous peroxidase was blocked for 10 min in 3% hydrogen peroxide and 10% methanol in PBS. Bound antibodies were visualized after washes in PBS by the avidin-biotin complex (VECTASTAIN® ABC-Peroxidase Kit; 1:250, PK-4000; Vector Laboratories, Servion, Switzerland) in combination with the DAB Substrate Kit (34002, ThermoFischer Scientific). The cultures were mounted with Aquatex (Millipore, Darmstadt, Germany).
For the analysis of mature neurons the cultures were stained for the general neuronal marker neuronal nuclei (NeuN). Cultures were treated as described above and incubated overnight at 4°C with the primary antibody mouse monoclonal anti-NeuN (1:500, Millipore, Zug, Switzerland). Subsequent to washes in PBS, the cultures were incubated for 2 h with the fluorescently labelled antibody Alexa® Fluor donkey anti-mouse 488 nm 1:250, Molecular Probes, Carlsbad, CA, USA). Cultures were then washed for 4 × 10 min in PBS and mounted in 0.1 M PBS containing 50% glycerol.
Histological Analyses
All cultures were analyzed under bright field illumination by a researcher blinded to the treatment groups as described by our group before 18 –22 . In brief: only cells with distinct immunoreactivity, clear neuronal shape, and visible neurites were counted as GABA-positive neurons. To obtain cell counts, 5.2% of the glass slide surface (113 mm2) in the culture dish was analyzed in six areas (in the upper, right, lower, left corners (distance 2.5 mm from edge) and twice in the center of the culture; each sized 0.97mm2) using an X10 objective in combination with an X10 ocular with counting grid (×100 magnification). If there was any doubt about the specification of GABA-ir neurons, the cell cultures were examined at a higher magnification (×400). Cell density was calculated per square millimeter (mean density in control cultures for GABA-ir neurons was 80.2 ± 5.8 cells per mm2). For the determination of GABA-ir cell densities after control and EPC-CM treatment alone, 85 and 79 cultures, respectively, were analyzed from nine independent experiments. For the analyses with drug treatments, 7–16 cultures per group were included from two to four independent experiments. Fluorescence pictures stained for NeuN (12 and 11 cultures from control and EPC-CM treatment, respectively; four independent experiments) were recorded using an Olympus epifluorescence microscope (BX51, Olympus, Tokyo, Japan) equipped with a digital camera (Olympus DP72, Olympus, Tokyo, Japan). NeuN-ir cell counts were analyzed in six areas (in the upper, right, lower, left corners (distance 2.5 mm from edge) and twice in the center of the culture; each sized 0.15 mm2) using an X20 objective (×200 magnification). Cell density was calculated per square millimeter (mean density in control cultures for NeuN-ir cells was 118.2 ± 5.6 cells per mm2).
Statistical Evaluation
For statistical analysis, a commercially available software package was used (Prism 7.04, GraphPad Software, La Jolla, CA, USA). To compare group means of several groups, one-way analysis of variance (ANOVA) followed by Newman-Keuls multiple comparison test was used. Statistical significance of two groups only was assessed by two-tailed unpaired t-test or by the non-parametric Mann-Whitney test based on the outcome of the D’Agostino & Pearson normality test. Differences were considered statistically significant at p < 0.05. Data are presented as mean + SEM.
Results
Effect of EPC-CM Treatment on GABA-ir Cell Densities in Dissociated GE Cultures
Cultures were grown for 7 DIV and exposed to the EPC-CM from DIV5–7. We observed that the density of GABA-ir neurons was significantly higher in EPC-CM-treated striatal cultures as compared to controls (by 41.4% ± 4.7%, p ≤ 0.0001) (Fig. 2). Similarly, the density of NeuN-ir neurons was significantly augmented after EPC-CM exposure (by 44.5 ± 17.7%, p ≤ 0.003) (Fig. 3). In line with these results, the striatal cultures treated with EPC-CM disclosed a significantly higher protein content compared to controls (by 1.33 fold, t2.4/14, p < 0.05) (Fig. 4A) as well as a slight but significant increase in number of viable cells (t2.07/46, p << 0.05) (Fig. 4B). EPC-CM treatment resulted in a slight increase in GFAP levels (by 1.3-fold); however, this did not reach statistical significance (Fig. 4C). Finally, EPC-CM administration from DIV5-7 induced significantly higher levels of PCNA (by 1.46-fold, t2.3/12, p < 0.05) (Fig. 4D).

Representative photomicrographs of GABA-ir neurons in fetal rat striatal cultures grown for 7 days in vitro (DIV) treated (A) without or (B) with endothelial progenitor cells secretome (EPC-CM) from DIV5-DIV7. Scale bars: 100 µm, 25 µm (insets).
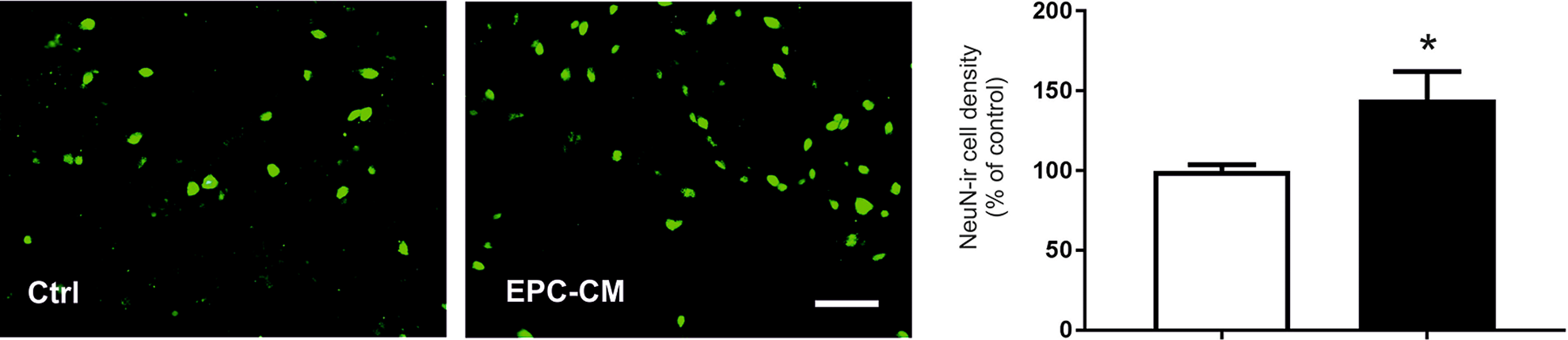
Representative photomicrographs and quantitative analysis of NeuN-ir neurons in fetal rat striatal cultures grown for 7 days in vitro (DIV) treated without (Ctrl, open bar) or with EPC-CM (filled bar) from DIV5 to DIV7. Scale bar: 50 µm. Data are presented as a percentage of controls and values given as mean + SEM. *p < 0.05.

Quantitative analysis of (A) total protein content, (B) viable cell number and (C, D) Western blot quantification of GFAP (C) and PCNA signals (D) in the striatal fetal cultures. Cultures were grown for 7 DIV and treated without (Ctrl) or with EPC-CM from DIV5 to DIV7. Data are presented as a percentage of controls and values given as mean + SEM. *p < 0.05 versus corresponding control.
EPC-CM Mediated Effects on GABA-ir Neurons Involve the ERK/MAPK and the AKT/PI3K Pathways
In our previous work, we reported that the ERK/MAPK and AKT/PI3K pathways are involved in mediating the angiogenic effects of EPC-CM on brain microvascular endothelial cells 4 . We therefore tested whether these pathways are implicated in the augmented GABA-ir cell densities observed after EPC-CM treatment. We found that the combined administration of the specific MEK/ERK inhibitor PD98059 resulted in a decrease in GABAergic cell densities as compared to EPC-CM treatment alone (F (3,52) = 5.45; p < 0.005) (Fig. 5A). Similarly, exposure of the cultures with the specific AKT/PI3K inhibitor LY294002 concomitantly with the EPC-CM treatment resulted in a reduction of GABAergic cell densities as compared to EPC-CM treatment alone (F (3,41) = 5.34; p < 0.005) (Fig. 5B). On the other hand, the ROCK pathway was not involved in the effects on GABA-ir cell densities observed after EPC-CM treatment. So, exposure of cultures with the selective ROCK inhibitor Y0503 and EPC-CM resulted in significantly higher as compared to controls (F (3,38) = 6.97; p < 0.001) (Fig. 5C).

Quantitative analysis of GABA-ir cell densities in the striatal fetal cultures. Cultures were grown for 7 DIV. The drugs PD98059 (PD; [10 µM]), LY294002 (LY; [10 µM]) and Y0503 (Y; [10 µM]) were added at DIV5–DIV7 concomitantly with EPC-CM. Untreated cultures served as controls (Ctrl). Note that the exposure to the inhibitors of the MEK/ERK (A) and the PI3K/AKT (B) pathways significantly reduced the EPC-CM induced higher GABAergic cell densities while the inhibitor of the ROCK pathway did not (C). Data are presented as a percentage of controls and values given as mean + SEM. *p < 0.05 versus corresponding controls.
EPC-CM Mediated Effects are Dependent on Translational Processes
In order to investigate whether translational processes were involved in the EPC-CM-induced higher GABA-ir cell densities, striatal cultures were exposed to the specific protein synthesis inhibitor cycloheximide (10 µM) from DIV5–7 in the absence or presence of EPC-CM. Inhibition of translation abolished the increase in densities of GABA-ir neurons after EPC-CM administration (F (3,22) = 16.04; p < 0.001) (Fig. 6).

Effects of EPC-CM treatment on cell densities of GABA-immunoreactive (-ir) neurons in striatal cultures exposed to cycloheximide. Cultures were grown for 7 DIV and exposed to cycloheximide [10 µM] from DIV5-7 in the absence (open bar) or presence of EPC-CM (filled bar). Untreated cultures served as controls (Ctrl). Note that the treatment with cycloheximide fully abolished the EPC-CM induced higher GABA-ir cell densities. Data are presented as a percentage of controls and values are given as mean + SEM. *p < 0.05 versus control.
Both Proteinaceous Factors and Lipidic Factors Contribute to the Increased GABA-ir Cell Numbers After EPC-CM Administration
EPC-CM contains a wide variety of growth factors and cytokines 4,5 . Hence, we aimed to assess whether the proteinase K treatment exerted an effect of the actions of EPC-CM. The higher GABA-ir cell densities induced by native EPC-CM was completely abrogated by the proteinase K treatment (F (3,31) = 15.25: p < 0.001) (Fig. 7A). Similar to the outcome seen for proteinase K treatment, removal of lipids from EPC-CM significantly diminished EPC-CM mediated effects on GABA-ir cell densities (F (3,28) = 5.47; p < 0.005) (Fig. 7B).

Effects of the protein content (by means of proteinase K digestion, prot-K; A) and of the lipids content (by means of chloroform extraction, chloro; B) from control medium (Ctrl) and EPC-CM on GABA-ir cell densities in fetal striatal cultures. Cultures were grown for 7 DIV and exposed to the modified EPC-CMs from DIV5 to DIV7. Note that both the inactivation of proteins and the removal of lipids of the EPC-CM resulted in significantly lower GABA-ir cell densities as compared to EPC-CM alone. Data are presented as a percentage of controls and values given as mean + SEM. *p < 0.05 versus corresponding controls; #p < 0.05 versus EPC-CM treated cultures.
EPC-CM Exerts Neuroprotection Against 3-NP-Induced Toxicity
To investigate if EPC-CM treatment exerts neuroprotective actions against an insult, cultures were exposed to the toxin 3-NP, alone or in combination with EPC-CM. As excepted, EPC-CM treatment significantly elevated GABA-ir cell densities, while exposure to the toxin 3NP significantly reduced GABA-ir cell densities as compared to controls (F (3,41) = 8.372; p < 0.001) (Fig. 8). Notably, the presence of EPC-CM during the insult resulted in a significant protection of the GABA-ir cell densities and presented as in controls (Fig. 8). EPC-CM exposure of cultures prior to the 3-NP treatment did not further alter the neuroprotective effects of EPC-CM on GABA-ir cell densities (Fig. 9A). In contrast, we detected that EPC-CM pretreatment resulted in significantly higher viable cell numbers as compared to the non-pretreated cultures after 3-NP exposure (F (2,30) = 12.52; p < 0.0001) (Fig. 9B).
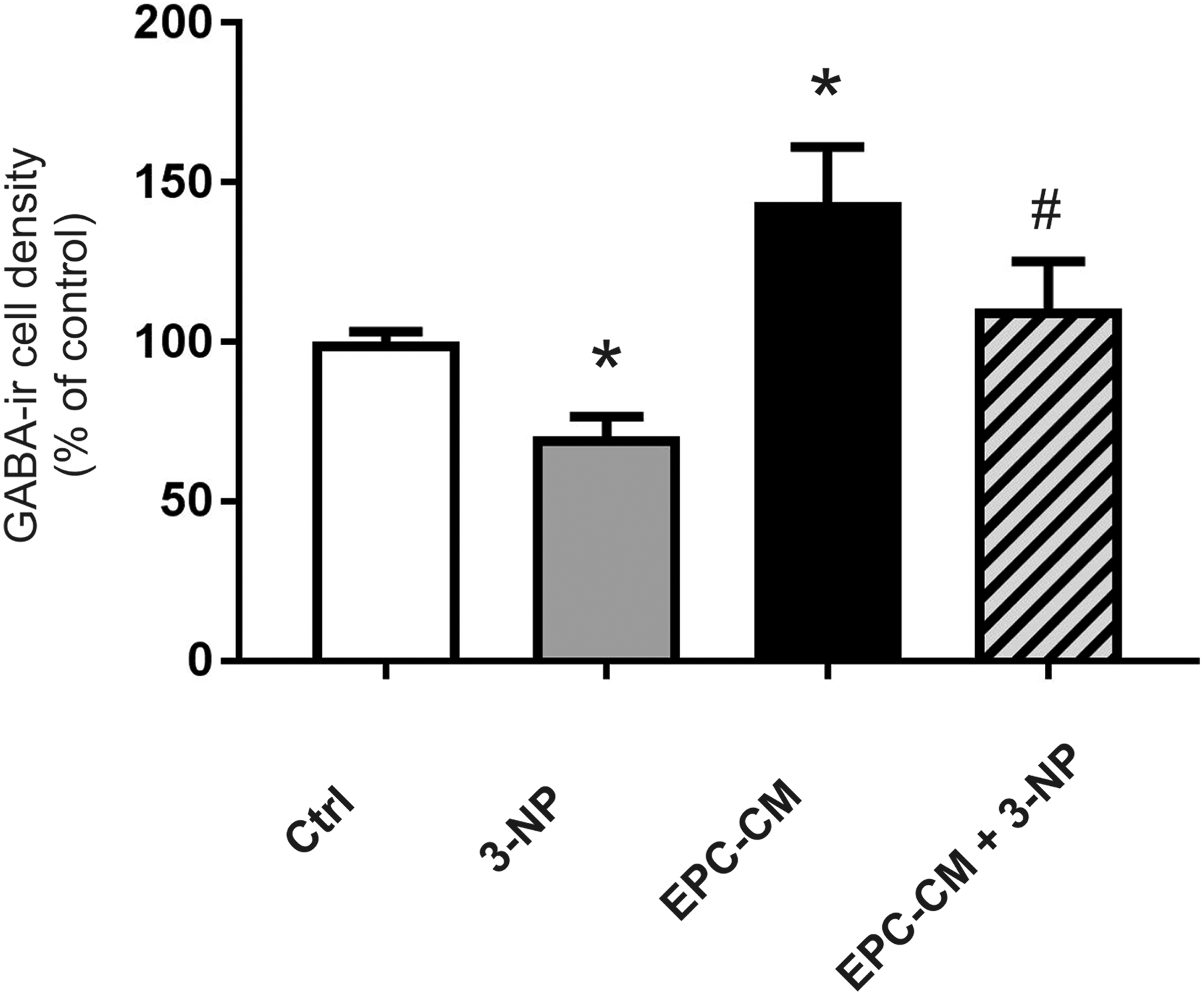
Quantitative analysis of GABA-ir cell densities in the striatal fetal cultures. Cultures were grown for 7 DIV. The drug 3-nitropropionic acid (3-NP, [5 mM]) was added at DIV5–DIV7 in absence (controls, Crtl) or presence of EPC-CM (EPC-CM + 3-NP). Data are presented as a percentage of controls and values are given as mean + SEM. *p < 0.05 versus control; #p < 0.05 versus 3-NP and EPC-CM treated cultures.
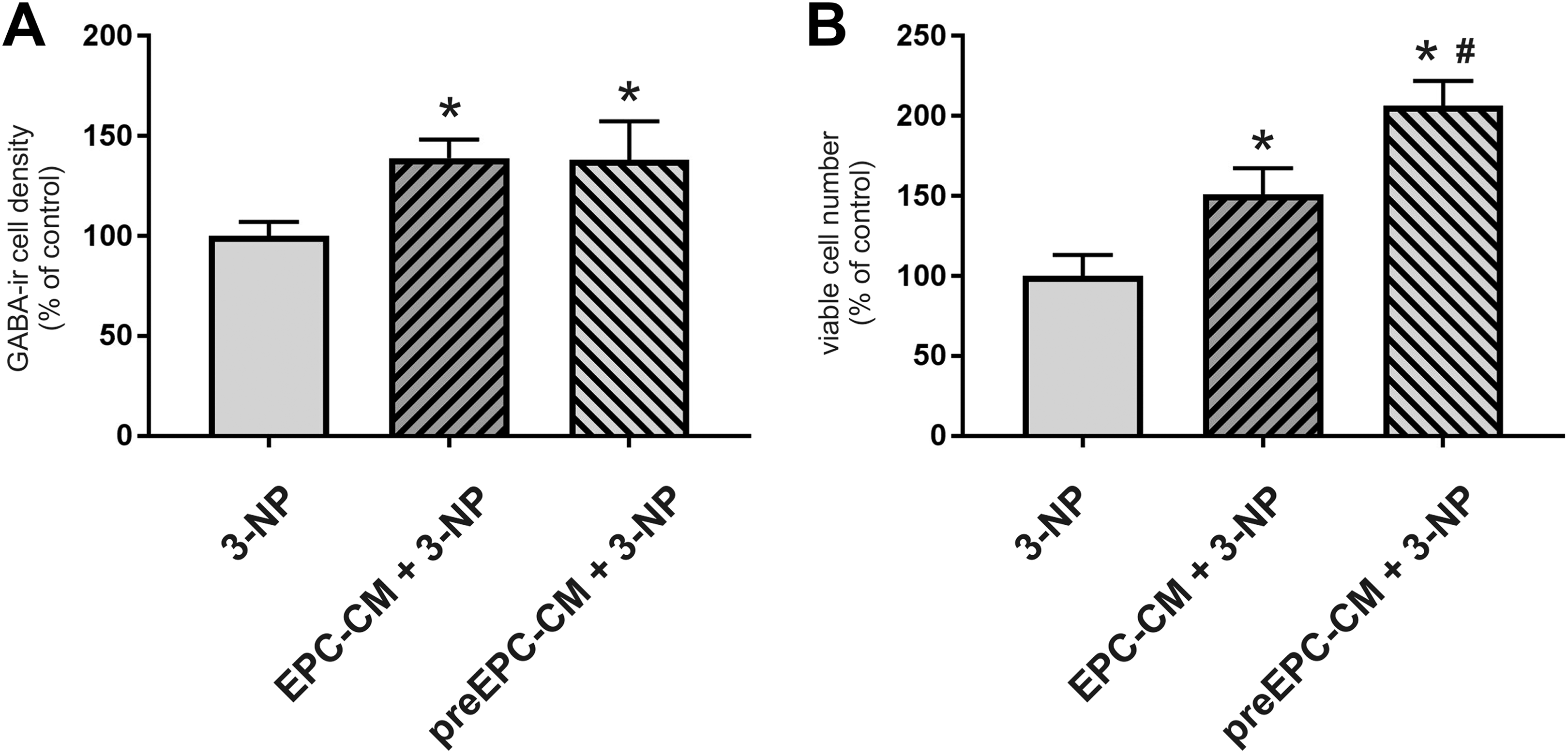
Quantitative analysis of GABA-ir cell densities (A) and viable cell numbers (B) in the striatal fetal cultures. Cultures were grown for 7 DIV. The drug 3-nitropropionic acid (3-NP, [5 mM]) was added at DIV5–DIV7 in absence (controls, Crtl) or presence of EPC-CM (EPC-CM + 3-NP). Cultures exposed to EPC-CM from DIV2 to DIV5 served as the pretreatment group (preEPC-CM + 3-NP). Data are presented as a percentage of controls and values are given as mean + SEM. *p < 0.05 versus 3-NP; #p < 0.05 versus EPC-CM + 3-NP treated cultures.
Discussion
The remarkable angiogenic capacity of EPC has been documented extensively 11,23 –25 , and these beneficial effects can be positively explained by the fact that the maintenance of a functional vascular network is essential for the delivery of nutrients, signaling molecules, and cells at the site of tissue injury. Observations made in the last decade suggest that EPC regenerative action might extend beyond vascular tissue 26 –28 . In line with this notion, there is mounting evidence that EPCs release a wide array of factors, including growth factors, cytokines, lipids, and extracellular matrix, which play a crucial role in supporting tissue viability and evoke a regenerative response 8,29 –31 . Currently, the concept of engraftment/transdifferentiation has been revisited, and the paracrine actions and induction of humoral effects in the host tissue are considered to be mainly responsible for the therapeutic effects of transplanted EPC 32 .
Preclinical studies have demonstrated that EPC paracrine factors, in the form of CM, can be exploited as a therapeutic tool (for a comprehensive review see 33 ); EPC-CM has been shown to relieve the ischemic injury in skeletal and myocardial muscle 3,31,34,35 . More recently, EPC-CM displayed promising neuroprotective potential for ischemic stroke 36,37 , promoted the dopaminergic phenotype in neuronal stem cells cultures 38 , and increased the number of doublecortin positive neuronal precursors in the subventricular zone of adult rats 39 . To the best of our knowledge, the present study is the first that specifically addresses the effects of EPC-CM on developing striatal cells, and attempts to characterize the cellular mechanisms involved.
The observed increase in densities of GABA-ir neurons in our experimental settings can be due to different mechanisms, i.e., a higher proliferation rate of neuronal progenitor cells and/or an increased survival and differentiation. That EPC-CM induced cell proliferation in our cultures is supported by the results of Western blot analysis for the general cell proliferation marker PCNA; however, it seems unlikely that this observation accounts for the increased GABAergic cell densities seen after the 2-day treatment. The finding that total protein content was significantly higher in the EPC-CM treated samples may hint to the notion that EPC-CM also increased numbers of other neuronal populations as well as other cell types. The former is corroborated by the observation of significantly higher numbers of NeuN-ir cells in EPC-CM treated cultures. In addition, Western blot analysis showed a tendency for GFAP to be higher in treated cultures, possibly also reflecting effects on glial cell populations. However, we did not specifically investigate these issues after the initial observations, but rather focused on characterizing the general factors, i.e. proteinaceous and lipidic, and signaling pathways involved in the EPC-CM-mediated increased densities of GABA-ir neurons.
In line with our previous observations in endothelial cells 4 , the response of striatal neurons to EPC-CM is modulated by activation of the PI3K/AKT and MAPK/ERK cascades. Interestingly, also creatine administration promoted GABAergic phenotype in striatal cultures through PI3K/AKT and MAPK/ERK 40 . The involvement of PI3K/AKT and MAPK/ERK in EPC-CM mediated effects is not surprising given the presence of several growth factors, e.g. brain-derived neurotrophic factor (BDNF) glial cell line-derived neurotrophic factor (GDNF), neuritin and vascular endothelial growth factor VEGF, known to be potent inducers of these signaling pathways 41 –44 . In the central nervous system AKT/PI3K signaling is important in regulating the survival and development of neurons. Furthermore, AKT/PI3K signaling provides neuroprotection against various stresses, including oxidative stress in the adult brain by blocking pro-apoptotic mechanisms (for review see Zhong 45 ) while ERK activation by small molecules exerts neuroprotection in vitro and in vivo models of HD 46 . Nevertheless, direct evidence by means of blocking antibody experiments for each of the potentially active neurotrophic factors mentioned above is necessary to prove their involvement in EPC-CM mediated effects. Inhibition of the ROCK pathway did not abolish the effects on GABA-ir cell densities after EPC-CM treatment. ROCK has been associated with neurite outgrowth, dendritic branching, and spine formation, which are all markedly inhibited when constitutively activated RhoA is expressed (for review see Amano et al. 47 ).
The observation that inhibition of translation by means of the protein translation inhibitor cycloheximide interfered with the actions of the EPC-CM may indicate that at least some of the effects were mediated indirectly, e.g. by production of growth factors acting in autocrine signaling. In line with this assumption, it has been reported that the translation of BDNF was increased in hippocampal neurons after activation by glutamate and BDNF itself and that this involved multiple signaling cascades, including RAS–ERK 48,49 .
The encouraging results concerning the use of stem cells secretomes on different clinical conditions has stimulated investigations aimed at the identification of the components of this vast collection of bioactive molecules. Proteomics studies have generated detailed maps of the peptides present in EPC-CM 29,31 ; however, comprehensive analyses of the population of lipidic factor released by these cells have not yet been performed. Though it is known that EPC release prostaglandins 50 , it can be excluded that this class of lipids contributed to the effect of EPC-CM described in our work since they are not extractable by chloroform 51 . Importantly, it has been reported that bone marrow progenitor cells (a class of cells that includes EPC), and mature endothelial cells, secrete sphingosine-1-phosphate (S1P) 52 , and its release is raised under hypoxic conditions. S1P is of particular interest as it is involved in neuronal differentiation through direct 53 and indirect mechanisms involving astrocytes 54 . However, at present, the possible presence and role of S1P in EPC-CM can only be speculated on. Moreover, it is now clear that proteins and lipids can be found in EPC-CM not only as free soluble molecules but also shuttled together with microRNA in exosomes and microvesicles 27,55 . Thus, taking in consideration the possible interactions among the different factors and their multiple modes of action, deciphering the overall secretome landscape will be a very complex task, although efforts in this direction have been taken for some exosomal cargoes 56 . In the present study, we have chosen to limit our investigation to the biochemical nature of the factors in EPC-CM able to modulate the yield of GABAergic cells. The significantly reduced GABA-ir cell densities following the proteolytic digestion and lipid removal treatments of EPC-CM demonstrates that both types of factors contribute to the EPC-CM effects. Lipids might serve as signaling molecules and energy substrates 57 , and which of the two functions were impaired by the treatments of EPC-CM was not addressed in our experimental setup. It cannot be excluded that chloroform treatment affected also the exosomes eventually present in our EPC-CM preparations.
Importantly, EPC-CM exerted neuroprotection against the irreversible succinate dehydrogenase inhibitor 3-NP—an observation that may be of importance in the context of neurodegenerative disorders with impaired energy metabolism due to a reduced mitochondrial complex-II and complex-III 58,59 . Interestingly, exposure to EPC-CM prior to the insult significantly augmented the cytoprotective effect on viable cell numbers, while there was no effect seen when assessing density of GABA-ir neurons. This likely reflects that EPC-CM mediated also cytoprotective actions for additional cells subpopulations in the striatal cultures. Support for this notion is given by the work of Fink and co-workers, who reported that 3-NP also shows toxicity for cultured hippocampal, septal, and hypothalamic neurons 60 .
In sum, the present work provides evidence that EPC-derived CM is able to directly support the survival of striatal progenitor cells. Moreover, our results show that these actions are mediated by lipidic and proteinaceous factors and involve the PI3K/AKT and MAPK/ERK signaling pathways. Importantly, we could demonstrate that EPC-CM is neuroprotective against 3-NP toxicity. In conclusion, our results suggest that EPC-CM might be useful to expand the repertoire of available tools to tackle neuronal degeneration.
Footnotes
Acknowledgments
The expert technical assistance of Susanne Wälchli is gratefully acknowledged. We thank Andrea Felser for the lipid measurements.
This work was presented in part at the 25th Annual Meetings of the American Society for Neural Therapy and Repair, Clearwater, USA (Cell Transpl. (2018); 27(4): 682).
Ethical Approval
All experiments were carried out in accordance with the guidelines of the Animal Research Ethics Committee of Canton Bern, Switzerland, and the University of Bern Animal Care and Use Committee, Switzerland (study No. 6/12 and 10/15).
Statement of Human and Animal Rights
All procedures in this study were conducted in accordance with the Animal Research Ethics Committee of the Canton Bern, Switzerland, and with approval of the University of Bern Animal Care and Use Committee, Switzerland (authorizations numbers BE6/12 and BE10/15).
Statement of Informed Consent
Informed consent for patient information to be published in this article was not obtained because the buffy coats employed for EPC isolation were purchased from the Interregional Transfusion Centre of the Swiss Red Cross (Bern, Switzerland) and no experimental approval or informed consent was needed.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Department of Neurosurgery, University of Bern, the HANELA Foundation and the Swiss National Science Foundation NRP63 (406340_128124).