
Abstract
MG132 is a pivotal inhibitor of the ubiquitin-proteasome system (UPS), and rapamycin (RAPA) is an important inducer of autophagy. MG132 and RAPA have been shown to be effective agents that can cure multiple autoimmune diseases by reducing inflammation. Although individual MG132 and RAPA showed protective effects for atherosclerosis (AS), the combined effect of these two drugs and its molecular mechanism are still unclear. In this article we investigate the regulation of oxidative modification of low-density lipoprotein (ox-LDL) stress and foam cell formation in the presence of both proteasome inhibitor MG132 and the autophagy inducer RAPA to uncover the molecular mechanism underlying this process. We established the foam cells model by ox-LDL and an animal model. Then, we tested six experimental groups of MG132, RAPA, and 3MA drugs. As a result, RAPA-induced autophagy reduces accumulation of polyubiquitinated proteins and apoptosis of foam cells. The combination of MG132 with RAPA not only suppressed expression of the inflammatory cytokines and formation of macrophage foam cells, but also significantly affected the NF-κB signaling pathway and the polarization of RAW 264.7 cells. These data suggest that the combination of proteasome inhibitor and autophagy inducer ameliorates the inflammatory response and reduces the formation of macrophage foam cells during development of AS. Our research provides a new way to suppress vascular inflammation and stabilize plaques of late atherosclerosis.
Keywords
Introduction
Atherosclerosis (AS) is a chronic inflammatory disease of humans, which lead blood vessels to becoming brittle and to age, clog, and harden 1 . Increased levels of oxidative modification on low-density lipoprotein (ox-LDL) is one of the important factors to induce or worsen AS. Ox-LDL-induced stress leads to the migration to the lesion area, and adhesion and aggregation of immune cells, especially macrophages, and causes cell damage. Present data demonstrate that macrophage recruitment of ox-LDL enhances production of surface factors and inflammatory mediators, including pro-inflammatory cytokines. Moreover, ox-LDL causes formation of lipid-laden macrophage foam cells in lesions via uptake of the scavenger receptor of macrophages. This robustly increases AS plaque and severity of disease 2 –4 . Thus, establishment of a macrophage-derived foam cell model is important for protection of macrophage cells from oxidative damage and treatment of AS.
The ubiquitin-proteasome system (UPS) is one of the major systems for degradation of proteins in cells and is closely correlated to proliferation, apoptosis, and metastasis of cells 5 . Evidence suggests that UPS plays an important role in regulation of the critical signaling pathways for atherosclerotic diseases, which involve vascular inflammation, apoptosis, oxidative stress, and cholesterol metabolism 6 .
Previous research identified that degradation of IkB-α by UPS leads to translocation of NF-κB to the nucleus and thus activation of cells 7 . Inflammatory factors such as TNF-α, IL-6, and IL-8 can be activated by the NF-κB pathway, causing AS 8,9 . However, UPS inhibitors are able to decrease the degradation of IkB-α and prevent NF-κB from activation in atherosclerotic plaques, thus suppressing inflammation of macrophages and endothelial cells 10,11 . In addition, UPS can rapidly degrade ABCA1, ABCG1, and apoB to cause lipid metabolism disorder and development of atherogenesis 12 –14 . UPS inhibitors such as MG132 increase the expression of LDLR and inhibitors prevent damage to lipid metabolism. 15 These studies demonstrate the importance of UPS inhibition for alleviation of atherosclerotic lesions and provide insight into effective AS therapy with proteasome inhibitors. However, there is growing evidence that indicates that suppression of UPS can lead to inhibition of the degradation of cell apoptosis proteins such as Bax and misfolding of proteins in the endoplasmic reticulum (ER), causing significant ER stress 16 . Moreover, massive and continuous expression of apoptotic proteins and deterioration of ER function could induce cell death 17 . Finally, apoptosis of foam cells of macrophages can develop a potentially fatal thrombosis in advanced AS 18 . It is therefore essential for advanced AS patients to reduce cell apoptosis in order to maintain atherosclerotic plaque stability.
Autophagy is another pathway of protein degradation in cells. Autophagy plays an important role in degradation of damaged organelles and proteins 19 , maintenance of the intracellular environment, and clearance of microbial infection. Studies show that UPS and autophagy are functionally compensated and that suppression of UPS will lead to activation of autophagy 17 . Whereas UPS rapidly degrades proteins, autophagy pathways selectively remove protein aggregates and damaged/excessive organelles, and play a protective function to mitigate ER stress caused by accumulation of the proteins 20 –22 . In AS, basal autophagy contributes to the degradation of ox-LDL, reduction of inflammatory cytokines, and inhibition of macrophage foam cell formation to stabilize AS plaque 23 –27 . Recently, it was reported that autophagy inducer RAPA selectively inhibited the PI3K/Akt/mTOR signaling pathway and regulated autophagy of macrophages and vulnerability of atherosclerotic plaque 28 . Thus, we hypothesize that autophagy plays a key role in the inhibition of atherosclerotic plaque formation when using proteasome inhibitors. To test this hypothesis, we first created a foam cell model and incited its inflammatory reaction by ox-LDL. Then, we verified the phenomena that UPS and autophagy are functionally coupled and that suppression of UPS activates autophagy in foam cells. In previous studies, we showed that autophagy attenuated ox-LDL stress-caused cell apoptosis and inflammation, and prevented foam cell formation, suggesting that autophagy is an essential element for the treatment of AS. In this study, we explored regulation of AS by both UPS and autophagy pathways and interrelationships between the two cellular degradation systems. We chose the autophagy inducer RAPA in combination with proteasome inhibitor MG132 to test the efficacy of the regulation of ox-LDL stress and foam cell formation for the first time 29 . We also established an animal model for studying the effect of drugs. The in vivo experiments with animals models were carried out in our laboratory. Our research provides a new way to suppress vascular inflammation and stabilize plaques in late AS. In addition, our studies may provide insight into other autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis.
Materials and Methods
Reagents and Antibodies
Reagents and antibodies used in this study are listed in Tables 1 and 2.
Reagents Used in This Study.

Antibodies Used in This Study.

High-Fat Diet-Induced AS in ApoE−/− Mice
Eight-week-old ApoE−/− mice (Nanjing Biomedical Research Institute, Nanjing, Jiangsu, China) were fed a high-fat diet (HFD) (Shoobree, Nanjing, Jiangsu, China) for 16 weeks to induce AS. Every effort was made to reduce animal suffering.
Atherosclerotic Lesion Analysis
Mice were euthanized and their hearts and aortas were isolated. Lesions were stained with Oil Red O (ORO; Sigma-Aldrich, St. Louis, MO, USA) for 30 min at room temperature (20–25°C) before being observed under a Stereo Microscope (OlympusSZ51, Tokyo, Japan). The aorta was opened longitudinally along the ventral midline from the iliac arteries to the aortic root. After the branching vessels were treated, the aorta was pinned flat on a black wax surface. Lesions were treated with 70% ethanol and then stained with Sudan IV for 15 min, washed with water for 10 min, and then stained with eosin for 3 min, destained with 80% ethanol, and then washed with phosphate-buffer saline (PBS) before being observed under the microscope.
Cell Culture and Foam Cell Induction
The RAW 264.7 cell line was obtained from the American Type Cell Culture Collection. Cells were maintained in Dulbecco’s modified eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a humidified atmosphere with 5% CO2. For pharmacological treatment, cells were cocultured with MG132 (10 μM), RAPA (200 nM), or 3-MA (5 μM) for 3 h and subsequently incubated with 40 μg/ml human ox-LDL for 24 h to induce foam cells before being harvested.
Cell Viability and Proliferation
The cytotoxicity of ox-LDL or drugs was analyzed using a Cell Counting Kit-8 (CCK8). In brief, the RAW 264.7 cells (1 × 104 cells/well) were plated on 96-well plates (Corning Incorporated, NY, USA). After incubation with drugs or ox-LDL for 24 h, 10 μl reagent was added to each well and further incubated for 1–4 h. The viability of cells was estimated by measurement of absorbance at 450 nm (A450) that was read with a microplate reader (INFINITE M200, Tecan, Mannedorf, Switzerland).
Cell apoptosis and necrosis were detected using an Annexin V-FITC/PI Kit in a flow cytometer based on published studies from our laboratory 30 (FACSCanto, BD Co. Inc., Franklin Lakes, NJ, USA).
ORO Staining and Cholesterol Measurement
Macrophage lipid accumulation and foam cell formation were examined by cholesterol measurements and ORO staining, respectively. RAW 264.7 cells were cultured in a six-well plate. Cells were treated with 40 μg/ml human ox-LDL for 24 h to induce foam cell formation when required. Cells were fixed in 4% paraformaldehyde for 20 min, and washed in PBS three times. Next, cell were stained with 0.5% ORO for 5 min at room temperature (20–25°C), and washed with water for 1 min. Then, cell were stained with hematoxylin for 1 min at room temperature (20–25°C), and washed with water for 3 min before being observed under the microscope. Cholesterol content was measured by cholesterol assay kit following the manufacturer’s instruction. Total and free cholesterol content were measured using a microplate reader. Cholesterol ester levels were calculated by normalization to protein levels for each sample.
Flow Cytometry Analysis
Fluorescent-activated cell sorting (FACS) analysis was performed with routine protocols using the FACSCalibur flow cytometer (BD Immunocytometry Systems, San Jose, CA, USA). Antibodies are listed in Table 2. RAW 264.7 cells were cultured in a six-well plate. After incubation with drugs for 3 h or ox-LDL for 24 h, cells were treated with Brefeldin A for 6 h before being harvested. Cell were washed with PBS and then stained and analyzed using a Fixation & Permeabilization kit, following the recommended protocols.
ELISA Assays
Cells were seeded in 96-well plates 3 h prior to drug treatment and then ox-LDL treatment 24 h. The concentration of NF-κB and iNOS was determined using ELISA kits according to the manufacturer’s instructions.
Western Blot Analysis
Cells were washed two or three times with ice-cold PBS and lysed in RIPA buffer containing protease and phosphatase inhibitors. Total protein in supernatant was measured using bicinchoninic acid (BCA) protein assay kit. Lysates of each sample were separated by 12% SDS-PAGE. Proteins in SDS−acrylamide gel were transferred to 0.22 or 0.45 μm PVDF membrane (Merck Millipore, Billerica, MA, USA) at 100 V for 30–60 min, fixed with 5% dry milk for 2 h at room temperature, and probed overnight with specific antibodies at 4°C. After washing three times for 10 min each with TBST (TBS + 0.1% Tween20), the blots were incubated with HRP-conjugated anti-IgG for 2 h at room temperature, detected on a Tanon instrument (Tanon 5500, Shanghai, China) by enhanced chemiluminescence. Quantification of band intensity was carried out using Image J software and were normalized to the β-actin levels.
EGFP-LC3B Transfections and Laser-Scanning Microscopy Observation
The RAW 264.7 cells were transfected with 2 μg EGFP-LC3B according to the manufacturer’s protocol. After incubation for 24–48 h, cells were treated with drugs for 3 h before being treated with ox-LDL at 40 mg/L for 24 h. Cells were observed by laser-scanning microscopy (LSM510/ ConfoCor2; Zeiss, Jena, Germany) with a 40× oil-immersion plan apochromat objective lens.
Fluorescent Microscope Observation
Autophagic vacuoles were detected by LysoTracker Red DND-99 (LTR). Cells were placed in Prolong Gold antifade with DAPI to stain nuclei. RAW 264.7 cells were incubated with drugs for 3 h before being treated with ox-LDL at 40 mg/L for 24 h, washed three times with PBS, incubated with the prewarmed (37°C) medium containing LTR for 1 h, and washed once with PBS. Cells were stained with DAPI and examined under a laser-scanning microscope (LSM510/ConfoCor2). Intensive red dots in the cytoplasm are representative of autophagic vacuoles.
Immunohistochemistry
The aortic arch tissues were placed in 10% formalin at room temperature overnight for paraffin embedding and were processed for immunohistochemical staining. Paraffin-embedded specimens were sectioned at 5 μm, deparaffinized, and hydrated in PBS. Then, the sections were incubated in 3% H2O2 for 10 min and rinsed with PBS. A primary antibody against cleaved caspase3 was applied, followed by washing and incubation in a biotinylated secondary antibody for 30 min. Last, tissues were observed under the microscope.
TUNEL Assay
The TUNEL method was performed to label the 3′-end of fragmented DNA of the apoptotic foam cells in the aortic arch after different treatments. The aortic arch treated as indicated were fixed with 10% neutral formalin liquid, then permeabilized by 0.1% Triton X-100 for FITC end-labeling the fragmented DNA of the apoptotic foam cells using a TUNEL apoptosis detection kit. The FITC-labeled TUNEL-positive cells were imaged using fluorescent microscopy at 488 nm excitation and 530 nm emission.
Statistical Analysis
Data were collected from three replicates per experiment. All data were analyzed by one-way analysis of variance (ANOVA) with post-Tukey’s post-test with the Graphpad Prism 6.0 software package: P < 0.05, P < 0.01, and P < 0.001 were considered as statistically significant differences. Bars in the graphs represent standard deviation (S.D.).
Results
Ox-LDL Converted RAW 264.7 to Macrophage Foam Cells and Changed Cytokine Polarization
RAW 264.7 cells share many features with macrophages and can be induced by ox-LDL to produce inflammatory cytokines and foam cells in vitro 31 . Expression of hallmark cytokine F4/80 confirmed the cell as a macrophage cell line (Fig. 1A). To find out the effective concentration of ox-LDL for inflammatory reaction and formation of foam cells, RAW 264.7 cells were treated with ox-LDL at various concentrations (0, 10, 20, 40, 60 mg/L) for 24 h and the level of inflammatory cytokines and foamy characterization were tested. The cytotoxicity of ox-LDL to RAW 264.7 cells was detected by using the CCK8 assay. Ox-LDL up to 40 mg/L is not toxic to RAW 264.7 cells (Fig. 1B, *P < 0.05). The Oil Red staining of the cells after treatment with ox-LDL at 40 mg/L for 24 h (Fig. 1C) showed that RAW 264.7 cells were effectively converted to foam cells. In addition, the assay with the total cholesterol reagent kit and free cholesterol kit revealed significant increase of the cholesterol esters level in the RAW 264.7 cells after stimulation by 40 mg/L ox-LDL for 24 h (Fig. 1D) (**P < 0.01; ***P < 0.001). These data collectively show that the RAW 264.7 cells can be effectively transformed into foam cells at 40 mg/L ox-LDL. Thus, 40 mg/L ox-LDL (24 h) is used in the subsequent experiments.

Ox-LDL converted RAW 264.7 to macrophage foam cells and changed cytokine polarization. (A) RAW 264.7 cells with or without ox-LDL were analyzed by FACS. Peak and histograms show the percentages of cells expressing F4/80. (B) RAW 264.7 cells were incubated with ox-LDL (0–60 μg/ml, 24 h), and CCK-8 assays were used to examine cell proliferation and viability. (C) Cells were exposed to different concentrations of ox-LDL (0–60 μg/ml, 24 h); cells were stained by Oil Red. Representative images show that ox-LDL induces foam cell formation. (D) Total cholesterol and free cholesterol measurement by cholesterol assay kit of RAW 264.7 cells treated with or without ox-LDL (0–60 μg/ml, 24 h). Cholesterol esters level is calculated for each cell group. (E) RAW 264.7 cells were cocultured with ox-LDL (0–60 μg/ml, 24h) and then analyzed by FACS. Surface and intracellular cytokines were used to detect the expression levels of polarization markers and pro-inflammatory cytokines. Data are the averages from three independent experiments. *P < 0.05, and **P < 0.01 versus corresponding controls.
To investigate the effect of ox-LDL on inflammatory cytokines, the RAW 264.7 cells were stimulated with 10–60 mg/ml ox-LDL for 24 h. Ox-LDL at 40 mg/L significantly increased the expression of inflammatory cytokines of IL-6 and TNF-α (Fig. 1E). However, the protein levels of IL-10 in the RAW 264.7 cells were lower than those of the control at 10–20 mg/L of ox-LDL, but higher at 40–60 mg/L. These data indicate that high concentrations of ox-LDL triggered the expression of anti-inflammatory cytokine IL-10, but low concentration of ox-LDL promoted expression of the pro-inflammatory cytokines IL-6 and TNF-α. In short, these data indicated that the 40 mg/L ox-LDL can trigger inflammation effectively.
To explore whether ox-LDL affects the polarization of macrophages, RAW 264.7 cells were examined by FACS for the hallmark cytokines of M1 and M2 polarization (Fig. 1E). FACS analysis showed that 10–40 mg/L ox-LDL stimulated RAW 264.7 cells to express higher levels of CD80, CD40, and CCR7, but caused little change in the CD206 expression. However, expression of all cytokines at 60 mg/L ox-LDL was lower than that of the 40 mg/L group, implying toxicity of high-concentration ox-LDL to RAW 264.7 cells. These results are comparable with the M1 phenotype, suggesting that ox-LDL promotes M1 polarization of RAW 264.7 cells.
Because HFD-induced arterial lesions in ApoE−/− mice have many features in common with human AS, ApoE−/− mice are considered to be a good animal model of AS 32 . We successfully established the ApoE−/− mice model of AS (Supplemental Fig. 1).
Autophagy Compensates UPS to Reduce Accumulation of Polyubiquitinated Proteins and Cell Apoptosis in Foam Cells
UPS and autophagy are functionally coupled, and suppression of UPS leads to activation of autophagy 33 . MG132 is a specific, potent, reversible, and cell-permeable proteasome inhibitor 34 . MG132 at concentration <10 µM showed no obvious cytotoxicity to foam cells within 1 h, but 10 µM MG132 showed low toxicity to foam cells after 3 h treatment (Fig. 2A). These result suggested that accumulation of apoptotic proteins and decompensation of ER function might induce foam cell death.
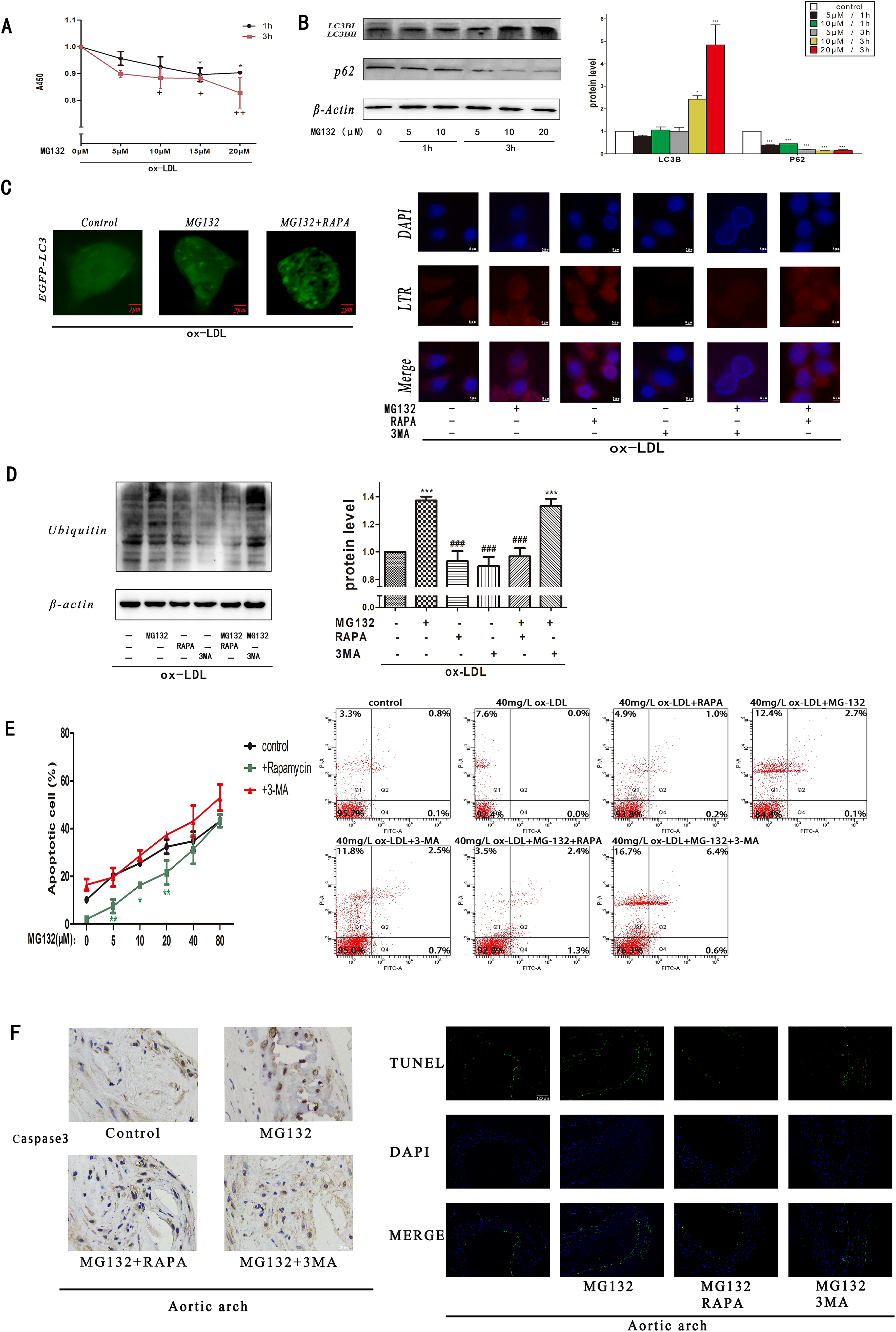
Autophagy compensates UPS to reduce accumulation of polyubiquitinated proteins and cell apoptosis in foam cells. (A) RAW 264.7 cells were incubated with MG132 (0–20 μM, 1–3 h) before treatment with ox-LDL (40 mg/L, 24 h). CCK-8 assays were used to examine cell proliferation and viability. (B) Immunoblots and densitometric analysis of autophagy-related proteins (LC3B and p62) normalized to β-actin treated with MG132 (0–20 μM, 1–3 h) before treatment with ox-LDL (40 mg/L, 24 h). (C) Fluorescence imaging of EGFP-LC3 on RAW 264.7 cells with or without treatment of MG132 (10 μM 3 h ) or RAPA (200 nM, 3 h) before treatment with ox-LDL (40mg/L, 24 h). The pictures were taken by confocal microscopy. Indication of autophagy in RAW 264.7 cells after incubating with different drugs (MG132, RAPA, and 3MA [5 μM]) for 3 h before treatment with ox-LDL at 40 mg/L for 24 h. Red dots represent autophagic lysosomes in RAW 264.7 cells. Blue dots represent nucleus in RAW 264.7 cells. (D) Immunoblots and densitometric analysis of ubiquitin proteins. It was normalized to RAW264.7 cells. Analysis of autophagy-related proteins (LC3B and p62) normalized to β-actin incubated with MG132 (0–80 μM, 3 h) with or without 3MA (5 mM) or RAPA (200 nM) treatment. CCK-8 assays were used to examine cell proliferation and viability. Survival rates of RAW 264.7 cells with or without treatment of MG132, 3MA, and RAPA were determined by Annexin V-FITC/PI staining and flow cytometry after treated with ox-LDL. (F) Immunohistochemistry of caspase-3 and TUNEL assay of the aortic arch tissues after different treatments. *P < 0.05, **P < 0.01, and ***P < 0.001 versus corresponding controls; +P < 0.05, and ++P < 0.01 versus corresponding controls;
In order to verify that inhibition of proteasome rapidly activates autophagy in foam cells, the levels of LC3B and p62 proteins were measured by immunoblot. We found that MG132 elicited accumulation of LC3B and degradation of p62 in a time- and dose-dependent manner (Fig. 2B). To test further, the EGFP-LC3B plasmid transfected cells for 48 h and the ability of proteasome inhibitor MG132 to induce autophagy in foam cells was measured. As shown in Fig. 2C, cells transfected with the EGFP-LC3B plasmid exhibited change of LC3B such that LC3BI diffuses in cytoplasm to condense into LC3BII as a bright spot after treatment with MG132 (10 µM) or MG132+RAPA (200 nM). These phenomena with the results that LC3BII expression increased in Fig 2B are conducted. Consistent with these findings, we used LysoTracker Red (LTR), which labels acidic compartments such as autophagic vacuoles or lysosomes and DAPI stains the nucleus of cells, to indirectly measure the ability of proteasome inhibitor MG132 to induce autophagy in foam cells. As shown in Fig. 2C, stimulation of the foam cells by MG132 (10 µM) or RAPA (200 nM) for 3 h increased the fluorescence intensity of LTR, but fluorescence from the 3MA (5 mM) groups was weak. These data strongly suggest the induction of autophagy by the inhibition of proteasome function in foam cells.
Based on the above findings, we reasoned that autophagy might play an important role when UPS is inhibited by MG132. To investigate the specific role of autophagy when UPS is inhibited, we tested whether autophagy inducer RAPA or autophagy inhibitor 3MA could affect the accumulation of polyubiquitinated proteins and cell apoptosis in foam cells. We found that MG132, but not 3MA, induced significant elevation of the polyubiquitinated proteins, as predicted (Fig. 2D). However, the treatment of MG132 in combination with RAPA significantly reduced the polyubiquitinated protein expression in comparison with the 3MA and MG132 treatment in foam cells. These results suggested that autophagy could be another pathway to degrade polyubiquitinated proteins.
We then investigated whether autophagy affects proteasome inhibitor-induced foam cell apoptosis by using both MG132 and 3MA. Interestingly, inhibition of autophagy by 3MA enhanced the apoptotic death of RAW 264.7 cells. However, apoptotic cell death was significantly reduced by the 3 h treatment of MG132 in combination with RAPA, as shown by the CCK8 assay (Fig. 2E). These results are similar to the observations from the flow cytometer experiments treated with ox-LDL. In short, these observations suggest that autophagy played a pro-survival role in proteasome inhibition in foam cells and that suppression of autophagy increased the sensitivity of cells to UPS inhibitor stimulation.
In addition, we have proved the above results in vivo. We assayed the level of caspase3 expression and TUNEL fluorescence intensity in aortic arch tissues that were treated with different drugs. We found the percentages of TUNEL-positive cells were increased in the MG132 group and MG132+3MA group compared with the control group. But following treatment with RAPA+MG132, the percentage of TUNEL-positive cells was decreased compared with the MG132 group and MG132+3MA group. The percentage of caspase3-positive cells exhibited a pattern that was similar to that of the TUNEL-positive cells mentioned above (Fig. 2F).
Combination of Proteasome Inhibitor with Autophagy Inducer Significantly Suppressed Expression of Inflammatory Cytokines and Formation of Macrophage Foam Cells
Previous research has shown that autophagy contributes to degradation of ox-LDL and decrease of inflammatory cytokines so as to relieve AS plaque 35 –37 . Thus, we hypothesized that combined use of autophagy and UPS generates synergistic effect on AS treatment. In order to explore this effect, we assayed the level of the inflammatory cytokines and characterized foam cells that were stimulated with ox-LDL and treated with autophagy inducer RAPA or autophagy inhibitor 3MA. FACS analysis showed that the MG132 and RAPA combination significantly suppressed cytoplasmic expression of IL-6 and TNF-α (Fig. 3A), and their expression levels are greatly decreased in comparison with the group of MG132 or RAPA alone. Moreover, the 3MA group significantly increased cytoplasmic expression of IL-6 and TNF-α. However, the protein expression level of IL-10 is opposite to those of IL6 and TNF-α. These results suggested that autophagy induction and UPS inhibition synergistically suppress the inflammatory factors of foam cells.
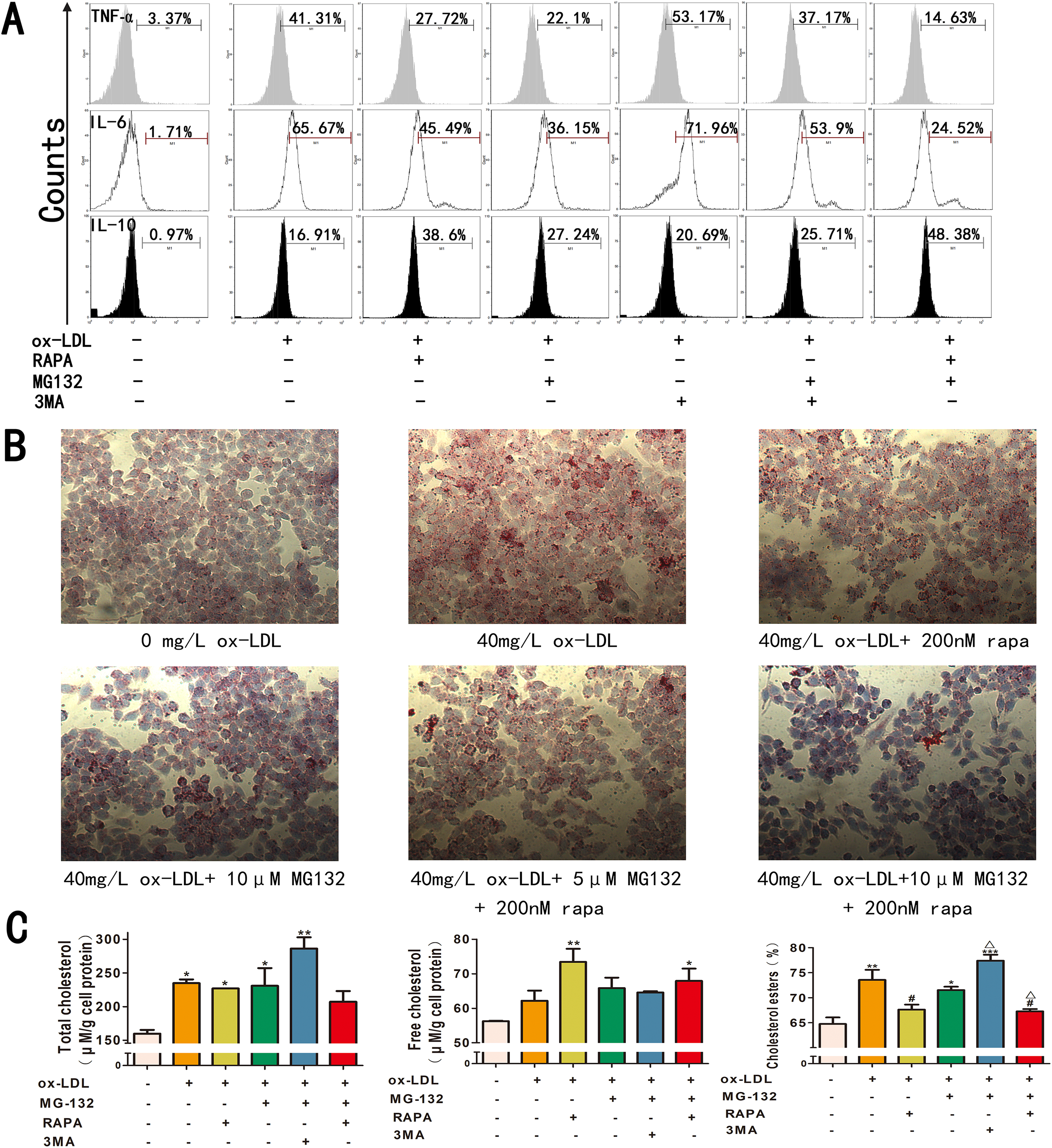
Combination of proteasome inhibitor with autophagy inducer significantly suppressed expression of inflammatory cytokines and formation of macrophage foam cells. (A) RAW 264.7 cells were cocultured with ox-LDL (24 h), stimulated by MG132, RAPA or 3MA (3 h), and then analyzed by FACS. Intracellular cytokine staining was used to detect the levels of pro-inflammatory cytokine expression. (B) Cells were stained by Oil Red. Representative images showed that MG132 and RAPA inhibited ox-LDL-induced foam cell formation. (C) Total cholesterol and free cholesterol are measured by cholesterol assay kit. Cholesterol esters level is calculated for each cell group. Data are the averages of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 versus corresponding controls;
To investigate the effect of the proteasome inhibitor and autophagy inducer on formation of macrophage foam cells, Oil Red staining was applied to the groups with the treatment of MG132, RAPA, or 3MA for 3 h. As shown in Fig. 3B, the proteasome inhibition in combination with autophagy significantly retarded the formation of macrophage foam cells. In addition, the cholesterol ester level is significantly reduced in the RAW 264.7 cells treated with both proteasome inhibitor and autophagy inducer (P < 0.05) (Fig. 3C). The effect of the combined RAPA and MG132 group is larger than that of the individual drug group. However, the cholesterol ester level of the combined 3MA and MG132 group is higher than those of all groups (P < 0.05). In summary, these data indicate that combination of proteasome inhibitor and autophagy inducer significantly suppress expression of inflammatory cytokines and formation of macrophage foam cells from RAW 264.7 cells. Based on this discovery, we further studied the mechanism in following experiments.
Combination of Proteasome Inhibitor with Autophagy Inducer Significantly Affects the NF-κB Pathway and the Polarization of Foam Cells
Given that the NF-κB pathway has been reported to be involved in AS progression 38,39 , we evaluated whether the NF-κB pathway is inhibited by the combined treatment of MG132 and RAPA. The ELISA assay was performed to examine NF-κB levels in the six cell culture groups. As shown in Fig. 4A, the ox-LDL group had a significantly higher NF-κB level than the control, and the combination of MG132 and RAPA had the lowest NF-κB level among all groups. In addition, we observed a decrease of IKB-α and increase of p-NF-κB after the ox-LDL treatment in comparison to the control (P < 0.001) (Fig. 4B), indicating activation of the NF-κB pathway. However, the MG132 and RAPA combination group has high expression of IKB-α in comparison with the ox-LDL group and the lowest p-NF-κB level. Finally, the protein level of p-JNK in the MG132 and RAPA combination group is higher than the RAPA group. In short, these data indicate that the inhibition of NF-κB but not JNK (c-Jun N-terminal kinase) pathway is intensified by the combined treatment of MG132 and RAPA.

Combination of proteasome inhibitor with autophagy inducer significantly affects the NF-κB pathway and the polarization of foam cells. (A) The amounts of NF-κB in the medium were measured using ELISA assay kits. (B) Immunoblots and densitometric analysis of inflammation-related proteins (IKB-α, p-NF-κB, and pJNK) normalized to β-actin. **P < 0.01 and ***P < 0.001 versus corresponding controls; #P < 0.05, ##P < 0.01, and ###P < 0.001 versus ox-LDL treatment. (C) RAW 264.7 cells were cocultured with ox-LDL (24 h), stimulated by MG132, RAPA, or 3MA (3 h), and then analyzed by FACS. Cytokine staining was used to detect the expression levels of polarization markers. (D) The amounts of iNOS in medium were measured using ELISA assay kits. ***P < 0.001 versus ox-LDL treatment.
Based on the earlier findings, we further explored whether the drugs affect the polarization of RAW 264.7 cells to decrease expression of inflammatory cytokines. Thus, RAW 264.7 cells were examined by FACS for polarization of the hallmark cytokines M1 and M2. FACS analysis showed that the expression of CD80, CD40, CCR7, and CD206 was significantly changed in the cells pre-stimulated with MG132, RAPA, or 3MA for 3 h before treatment with 40mg/L ox-LDL (Fig. 4C). The expression level of cytokines CD80 and CD40 of the ox-LDL group is higher than that of the MG132 and RAPA combination group, but lower than the RAPA group. In addition, the MG132 and RAPA combination group showed higher expression level of cytokine CCR7 than the ox-LDL group, but lower than the MG132 group. The expression level of cytokine CD206 in the MG132 and RAPA combination group is much higher than those of the ox-LDL and RAPA groups. In addition, an ELISA assay was performed to examine iNOS levels, which is a classical M1 macrophage marker in the five cell culture groups. As shown in Fig. 4D, the ox-LDL group has significantly higher iNOS level than the control, and the combination of MG132 and RAPA has a lower iNOS level than the individual drugs. Taken together, these data indicate that the combined treatment of MG132 and RAPA effectively suppressed M1 polarization of RAW 264.7 cells from M2-type macrophages, in comparison with the single-drug treatment groups. These results suggest that the M1 to M2 polarization of RAW 264.7 cells transformed immune response to immune tolerance so as to decrease inflammatory cytokine expression.
Discussion
Change of the Polarity May be a New Direction for Treatment of AS
Macrophages are divided into two types, M1 and M2, respectively representing immune response and immune tolerance 40 . INOS, CD40, CD80, and CCR7 are often the markers of M1 macrophages, and CD206 is the marker of M2 macrophages 41,42 . Earlier studies have showed that the polarization of macrophages is reversible in AS 43 . Macrophages can induce inflammatory injury when M2-types are transformed into M1-types in AS. Suppressing M1 polarization of foam cells can prevent inflammation and stabilize atherosclerotic plaque 44,45 . Our experiments demonstrated that the polarization caused by ox-LDL transformed RAW 264.7 to M1-type (Fig. 1E). The polar transformation of RAW 264.7 may be the reason why ox-LDL triggers inflammation in AS. We found that RAPA reduced the protein expression of CCR7, and is opposite to the effect of MG132. On the other hand, MG132, but not RAPA, reduced the protein expression of CD40 and CD80. The combined use of MG132 and RAPA increased expression of CD206, an M2 macrophage marker, but reduced expression of the M1 macrophage markers of iNOS, CD40, CD80, and CCR7 (Fig. 4C,D). The transformation of M1 into M2 macrophages by the combined group is more effective than any single drug. Although explanation of the cell transformation from immune response of M1 into immune tolerance of M2 requires further experiments, the combination of proteasome inhibitor with autophagy inducer might potentially represent a new direction for treatment of AS.
Autophagy May Play a Role in Stabilizing Foam Cells in the Presence of Proteasome Inhibitor MG132
Although UPS and autophagy are generally considered to be independent of each other in terms of the degradation mechanisms and objects 46 , growing evidence shows their connection 47 –49 . Suppression of UPS will lead to activation of autophagy, while suppression of autophagy enhances proteasome inhibitor-induced accumulation of polyubiquitinated proteins and cell death 17 . However, reduction of polyubiquitinated proteins and cell death by UPS inhibition and autophagy activation is often limited. We thought that autophagy inducer such as RAPA could significantly reduce the accumulation of polyubiquitinated proteins and cell death, and that autophagy serves as the compensation mechanism to UPS in foam cells, as supported by our experiments (Fig. 2). In short, the combination of MG132 and RAPA reduced the side effects such as accumulation of polyubiquitinated proteins and cell death caused by using MG132 alone, and remission of cell apoptosis in late-stage AS. Ultimately, it can play a stabilizing role in the atherosclerotic plaque.
The Mechanism for Regulation on Foam Cell Inflammation by Combined Use of MG132 and RAPA
Studies have showed that the UPS inhibitor MG132 prevented IKB-α degradation to control the activity of NF-κB 50,51 , and the autophagy activator RAPA affected multiple pathways such as NF-κB and JNK pathways and inhibited the inflammatory response 24,52 . Our experiments using ELISA and Western Blot revealed involvement of the key proteins in the NF-κB pathway. On the other hand, our results showed that the combination of RAPA and MG132 has higher pJNK protein activity than RAPA alone (Fig. 4). Since proteasome inhibitor MG132 does not decrease the activity of pJNK proteins, it may imply that MG132 targets further downstream than the JNK pathway to reduce inflammation. In short, reduction of cell inflammation by the combination of drugs may be achieved via the NF-κB pathway, but not the JNK pathway. In addition, the changes of cell polarity caused by the drug combination may be a factor affecting cell inflammation and foam cell formation.
In summary, combination of proteasome inhibitor with autophagy inducer effectively stabilized AS plaque, reduced cell inflammation, and changed the polarity of foam cells. We are also carrying out some scientific experiments in vivo using animals models to confirm these discoveries.
Conclusions
In short, the combination of MG132 and RAPA reduced cell death caused by using MG132 alone and remission in cell apoptosis at the aortic arch. Our studies also suggest that combination of the proteasome inhibitor MG132 with autophagy inducer RAPA ameliorates the inflammatory response and reduces the formation of macrophage foam cells (Fig. 5). This regulation is achieved via inhibiting the NF-κB pathways and affecting the polarization of RAW 264.7 cells.

A hypothetical schema on relation between cell polarization, UPS, and autophagy.
Supplemental Material
Supplementary_figure - Interplay of Autophagy Inducer Rapamycin and Proteasome Inhibitor MG132 in Reduction of Foam Cell Formation and Inflammatory Cytokine Expression
Supplementary_figure for Interplay of Autophagy Inducer Rapamycin and Proteasome Inhibitor MG132 in Reduction of Foam Cell Formation and Inflammatory Cytokine Expression by Wei Zhang, Wan Xu, Wenli Chen, and Quan Zhou in Cell Transplantation
Footnotes
Acknowledgments
We are grateful to Dr. Yoshimori (Laboratory of Genetics Graduate School of Medicine, Osaka University) for kindly providing the plasmid EGFP-LC3. We would like to thank all participants of this study and acknowledge invaluable support of Professor Hengming Ke, UNC-Chapel Hill School of Medicine of USA, for proofreading the manuscript. Sincere thanks go to every lovely and kind one in my life.
Ethical Approval
All experiments were conducted according to the Code of Ethics of the World Medical Association (Declaration of Helsinki), and all experimental protocols involving animals were approved by the Animal Care and Use Committee of Guangdong Province, China.
Statement of Human and Animal Rights
All procedures in this study were conducted in accordance with the Animal Care and Use Committee of Guangdong Province, China approved protocols.
Statement of Informed Consent
Statement of Informed Consent is not applicable.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: This study was supported by the National Natural Science Foundation of China (81471659 and 81630046) (Department of Radiology, The Third Affiliated Hospital of Southern Medical University, Guangzhou 510630, China). This research is supported by a grant from the Science and Technology Project of Guangzhou (Grant No. 201805010002).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.