
Abstract
The interaction of SDF-1α (also known as CXCL12) with the CXCR4 receptor plays a critical role in the retention of hematopoietic stem cells (HSCs) in bone marrow. The viral macrophage inflammatory protein-II (vMIP-II), a human herpesvirus-8 (HHV-8)-encoded viral chemokine, can bind the CXCR4 receptor and inhibit endogenous ligand-induced calcium responses and cell migration. Previously, we used the bivalent ligand approach to link synthetically two unnatural D-amino acid peptides derived from the N-terminus of vMIP-II (DV1 and DV3, respectively) to generate a dimeric peptide, DV1-K-(DV3) (also named HC4319), which shows very high affinity for CXCR4. Here, we studied the biological effects of this dimeric peptide, HC4319, and its monomeric counterpart, DV1, on SDF-1α-induced signaling in CXCR4- or CXCR7-transfected Chinese hamster ovary cells and mobilization of hematopoietic progenitor cells (HPCs) in C3H/HeJ mice using an HPC assay. HC4319 and DV1 inhibited significantly the phosphorylation of Akt and Erk, known to be downstream signaling events of CXCR4. This in vivo study in C3H/HeJ mice showed that HC4319 and DV-1 strongly induced rapid mobilization of granulocyte–macrophage colony-forming units (CFUs), erythrocyte burst-forming units, and granulocyte–erythrocyte–monocyte–megakaryocyte CFUs from the bone marrow to the blood. These results provide the first reported experimental evidence that bivalent and D-amino acid peptides derived from the N-terminus of vMIP-II are potent mobilizers of HPCs in C3H/HeJ mice and support the further development of such agents for clinical application.
Introduction
Hematopoietic stem cells (HSCs) have the ability to produce multipotential and lineage-committed progenitor cells and blood cells. Under normal conditions, hematopoiesis occurs mainly in the bone marrow, so the peripheral blood typically contains only a few HSCs and hematopoietic progenitor cells (HPCs). However, HSCs and HPCs can be mobilized from the bone marrow into the circulation in response to certain cytokines (e.g., granulocyte colony-stimulating factor), myelosuppressive anti-tumor drugs, or CXCR4 antagonists such as AMD3100, which block the interaction between CXCR4 on HSCs and SDF-1α on bone marrow stromal cells.
CXCR4 belongs to the G-protein-coupled receptor superfamily and is a member of the CXC chemokine receptor family. It recognizes a specific ligand, SDF-1α (CXCL12). Such binding increases the phosphorylation of CXCR4 downstream signaling molecules such as Erk1/2 kinases, Akt, and nuclear factor-kappa beta 1 . CXCR4 plays a critical role in cell migration and survival and in the proliferation and retention of HSCs in the bone marrow 2 –5 . Clinical studies have shown that blockade of the interaction of CXCR4 and SDF-1α by a CXCR4 antagonist, AMD3100, results in rapid mobilization of HSCs to the peripheral blood 6 .
Viral macrophage inflammatory protein-II (vMIP-II) is a viral CC chemokine encoded by the human herpesvirus-8 (HHV-8) virus. It binds to a variety of chemokine receptors, including CXCR4, and acts as an antagonist or agonist to modulate the migration of different immune cell subsets for selective recruitment of Th2 cells and evasion from cell-mediated immunity 7 –12 . The inhibitory properties of vMIP-II on proinflammatory chemokine receptors has led to investigations on its ability to reduce the lymphocyte infiltration and Th1-cell-driven inflammatory responses in mice with spinal cord contusion injury, focal cerebral ischemia, or allografts 13 –16 . Under pathological conditions, vMIP-II also binds to CXCR4 or CCR5, which are HIV entry co-receptors, thereby moderating the inhibitory effect on HIV-1 entry 17,18 . In a CXCR4-transfected cell line, vMIP-II inhibits endogenous CXCR4 agonist-induced calcium responses and cell migration.
Previously, we designed a series of vMIP-II-derived CXCR4 antagonists and demonstrated that the N-terminus of vMIP-II was the critical sequence for binding to the CXCR4 receptor. Determination of the crystal structure of vMIP-II in complex with CXCR4 and structure–function studies of peptides derived from N-terminus of vMIP-II confirmed the importance of the N-terminus of vMIP-II 19,20 . We also found a remarkable stereochemical flexibility of the interface between CXCR4 and peptide inhibitors based on the observation that both D- and L-peptides derived from the same amino acid sequence of the N-terminus of vMIP-II showed strong and specific binding for CXCR4 and potently inhibited the entry and infection of the HIV strains for this particular co-receptor 21 .
We also reported previously a novel vMIP-II-derived bivalent antagonist, DV1-K-(DV3), which is designated as HC4319 hereafter and contains two CXCR4 monomeric ligands, DV1 and DV3, linked chemically via the side chain of a D-Lys residue 22 . Both DV1 and DV3 are D-amino acid peptides derived from the N-terminus (residues 1–21 and 1–10, respectively) of vMIP-II and have CXCR4-binding affinity and anti-HIV entry activity. The synthetic linkage of these two monomeric peptides led to the bivalent, dimeric peptide ligand HC4319, which has much higher CXCR4 affinity and biological activity than the monomeric peptides.
In this study, we investigated the effect of HC4319 and DV1 on SDF-1α-induced signaling in CXCR4- or CXCR7-transfected Chinese hamster ovary (CHO) cells. We also performed the first reported evaluation of the effect of peptides derived from the N-terminus of vMIP-II on the mobilization of HPCs in C3H/HeJ mice using a colony-forming unit (CFU) assay. CXCR7 (RDC1) is the second identified receptor of SDF-1α and its affinity to SDF-1α is higher than that of SDF-1α to CXCR4 23,24 . Therefore, we investigated whether CXCR7 is involved in the phosphorylation of Akt and Erk and if vMIP-II-derived peptides can also bind CXCR7.
Materials and Methods
Peptide Synthesis
DV1 and HC4319 were prepared by 9-fluorenylmethoxy carbonyl (Fmoc) chemistry and manual solid-phase peptide synthesis using a TentaGel S RAM (0.24 mmol/g) resin as the solid support. A 5-fold excess of Na-Fmoc-amino acid, diisopropyl-carbodiimide, and hydroxybenzotriazole was used in every coupling reaction step. Removal of the N-terminal Fmoc group was accomplished by 20% piperidine in dimethylformamide (DMF) for two cycles (5 min and 15 min, respectively). The Dde (N-[1-(4, 4-dimethyl-2, 6-dioxocyclohex-1-ylidene) ethyl]) group was de-protected with 2% hydrazine in DMF for 2 min and the step was repeated three times. The synthesis of HC4319 was started from a DV3/DV1 peptide with a C-terminal Lys. The second DV3 moiety was added from the C-terminus to N-terminus by coupling it through the ∊-amine group of the Lys side chain. The cleavage of the peptide from the resin was conducted with a cleavage cocktail containing water (5%, vol/vol), thiophenol (5%, vol/vol), and trifluoroacetic acid (90%, vol/vol) for 2 h at room temperature with gentle stirring. The peptide was precipitated by adding ice-cold diethyl ether and washed repeatedly in cold diethyl ether. The crude peptide was dissolved in 20% acetonitrile in deionized water before lyophilization and purified using semi-preparative reverse-phase high-performance liquid chromatography (RP-HPLC). The fractions containing the peptide were pooled together and lyophilized. The purity of the final product was assessed by analytical RP-HPLC and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. The purity of all peptides was at least 95%.
Transfection of Adherent CHO Cells
The plasmids pcDNA3-hCXCR4 and pcDNA3-hCXCR7 were stably transfected into CHO cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. A selective medium containing G418 (1.2 mg/mL) was used to isolate stably transfected cells, which were subsequently singly cloned. CHO cells expressing CXCR4 were obtained by fluorescence-activated cell sorting (FACS) using a BD FACSAria III instrument (BD Biosciences, San Jose, CA, USA).
CXCR7 Competitive Binding Assays Using a Labeled Antibody
CXCR7-transfected CHO cells were cultured in Dulbecco’s modified Eagle’s medium with 10% (v/v) fetal bovine serum, 100 IU penicillin, 0.1 mg/mL streptomycin, and 2 mM L-glutamine. G418 (400 μg/mL) was used as an agent to select CHO cells that stably expressed CXCR7. The transfected cells were washed twice with FACS buffer [0.5% bovine serum albumin (BSA), 0.05% sodium azide in phosphate-buffered saline (PBS)] prior to the binding assay. The cells were then seeded in V-shaped 96-well plates at 5 × 105 cells/well and incubated with various concentrations of DV1 or HC4319 and primary antibody (1:3000, mouse anti-human CD184 antibody, BD Biosciences) for 40 min on ice. After two washes with assay buffer, cells were incubated with secondary antibody (1:250, anti-mouse IgG-FITC antibody, Sigma-Aldrich, St. Louis, MO, USA) for 30 min on ice. After two washes by centrifugation with assay buffer, 50 μL of FACS buffer was added to each well. The fluorescence (485EX/528EM) value was recorded using a Synergy 2 microplate reader (BioTek Instruments, Winooski, VT, USA) and expressed as a percentage of the control group values. The competitive binding assays were performed in duplicate and the results are presented as mean ± standard error of the mean.
Flow Cytometric Analysis
CHO cells transfected with either CXCR4 or CXCR7 and wild-type CHO cells were washed with PBS containing 1% BSA and then stained at 4°C for 20 min with monoclonal anti-CXCR4 or anti-CXCR7 antibodies. The stained cells were analyzed by flow cytometry on an Accuri C6 flow cytometer (BD Biosciences). Data were analyzed using FlowJo Software (TreeStar Inc., Ashland, OR, USA).
Western Blotting
Cells were lysed in lysis buffer [100 mM NaCl, 50 mM Tris-HCl, 1 mM EGTA, 0.5% sodium deoxycholate, 1% Triton X-100, and 0.2% sodium dodecyl sulfate (SDS), pH 7.2] in the presence of phosphatase and protease inhibitor cocktail (Pierce, IL, USA) for 30 min on ice. Cell lysates were centrifuged at 17,000 g for 15 min at 4°C and the protein concentrations of the supernatants were determined with a BCA assay (Pierce). Equal amounts of protein mixed with 5× SDS loading buffer were loaded and separated on 8% SDS-PAGE gels and then transferred to nitrocellulose membranes (GE Healthcare, NJ, USA). After blocking with 5% milk or 7.5% BSA in Tris-buffered saline plus Tween-20 (TBST) (0.1% Tween-20 in TBS), the membranes were incubated with primary antibodies overnight at 4°C, washed three times in TBST, incubated with horseradish peroxidase (HRP)-coupled secondary antibodies for 1 hour at room temperature, and washed three times in TBST. The immunostained proteins were detected using a chemiluminescent HRP detection substrate (Pierce).
Colony-Forming Assays
All animal experiments were approved by the Laboratory Animal Research Center of Tsinghua University. C3H/HeJ Mice were administered subcutaneously with a vehicle control or the peptides. After 1 hour, peripheral blood was collected into heparin-treated tubes and peripheral blood cells were obtained after lysis with ammonium chloride solution (StemCell, Canada). Cells were cultured in MethoCult GF M3434 (StemCell, Canada) for 7 days under a humidified atmosphere with 5% CO2 and granulocyte–macrophage CFUs (CFU-GMs), erythrocyte burst-forming units (BFU-Es), and granulocyte–erythrocyte–monocyte–megakaryocyte CFUs (CFU-GEMMs) were counted.
Results
Enhanced Inhibitory Effect of Bivalent Peptide on CXCR4 Signaling
We determined the effect of vMIP-II-derived peptides on signaling transduction through the CXCR4 receptor by stably transfecting human CXCR4 into CHO cells and detecting the phosphorylation of Akt and Erk. Approximately 95% of CHO cells were positive for CXCR4 after transfection (Fig. 1A). SDF-1α (12.5 nM) induced the phosphorylation of Akt and Erk significantly and both DV1 (30 μM) and HC4319 (30 μM) inhibited the phosphorylation of Akt and Erk markedly. Compared with the monovalent peptide DV1, the bivalent peptide HC4319 has more potent inhibitory effect on the phosphorylation of Akt and Erk (Fig. 1B). These results demonstrated that vMIP-II-derived peptides can inhibit the phosphorylation of Akt and Erk induced by SDF-1α significantly.

Inhibitory effect of vMIP-II-derived peptides on SDF-1α-induced phosphorylation of signaling molecules in CXCR4-transfected CHO cells. (A) Expression of CXCR4 by the infected CHO cells analyzed by flow cytometry. (B) Effect of HC4319 and DV1 on SDF-1α-induced phosphorylation of Akt and Erk in CXCR4-transfected CHO cells detected by Western blotting.
Weak Inhibitory Effect of vMIP-II-Derived Peptides on CXCR7-Mediated Akt Phosphorylation
To determine whether the above observed phosphorylation was possibly due to CXCR7, which is known to be the alternative receptor of SDF-1α, we constructed CXCR7-transfected CHO cells and examined the phosphorylation of Akt and Erk at 5, 15, 30, 45, and 60 minutes in the presence of SDF-1α. Approximately 95% of CHO cells were positive for CXCR7 after transfection (Fig. 2A). At 5, 15, 30, 45, and 60 minutes after SDF-1α (12.5 nM) treatment, only Akt phosphorylation was observed consistently from 5 to 60 minutes; no phosphorylation changes were observed for Erk in CXCR7-transfected CHO cells (Fig. 2B). We further determined that Akt phosphorylation was induced through the transfected CXCR7 receptor and not by any other preexisting receptors on the CHO cells by examining the phosphorylation changes in Akt in wild-type CHO cells treated with SDF-1α (12.5 nM). We found no changes in Akt phosphorylation in the wild-type CHO cells (Fig. 2C) in response to SDF-1α, which confirmed that the Akt phosphorylation observed in CXCR7-transfected cells was mediated through the CXCR7 receptor. We also examined the effect of vMIP-II-derived peptides on the Akt phosphorylation induced by SDF-1α in these CXCR7-transfected CHO cells. Only DV1 had a slight effect on the SDF-1α-induced Akt phosphorylation in CXCR7-transfected cells (Fig. 2D). Therefore, SDF-1α could induce Akt phosphorylation, but had no effect on the phosphorylation of Erk in CXCR7-transfected CHO cells. vMIP-II-derived peptides had almost no effect on the Akt phosphorylation induced by SDF-1α in these cells.
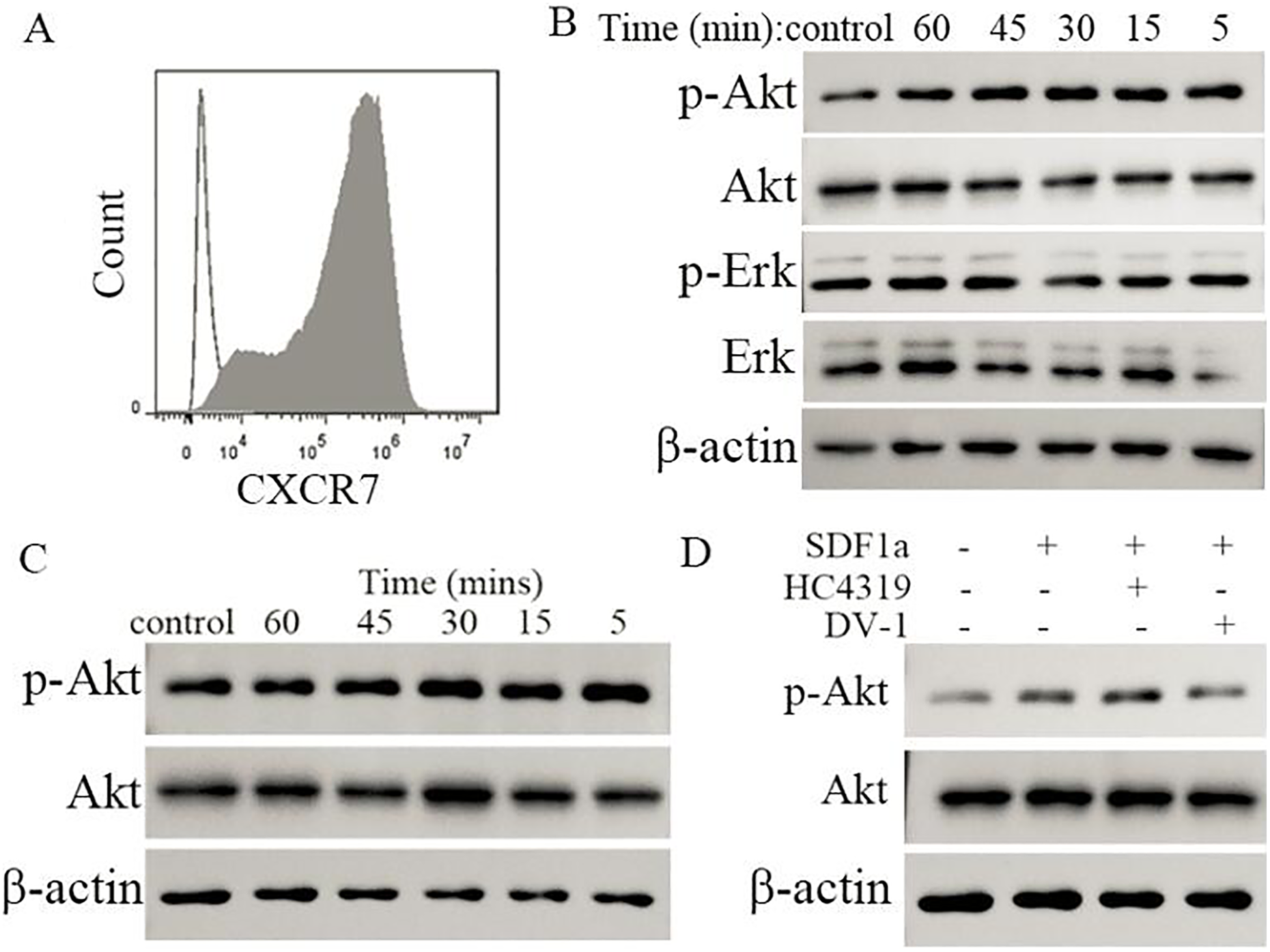
Weak phosphorylation of Akt induced by SDF-1α through the CXCR7 receptor and little inhibitory effects on CXCR7-mediated Akt phosphorylation by HC4319 and DV-1. (A) Expression of CXCR7 on infected CHO cells analyzed by flow cytometry. (B) Effects of SDF-1α on the phosphorylation of Akt and Erk in CXCR7high CHO cells detected by Western blotting. (C) Effects of SDF-1α on the phosphorylation of Akt in wild-type CHO cells detected by Western blotting. (D) Effects of HC4319 and DV-1 on SDF-1α-induced phosphorylation of Akt in CXCR7high CHO cells assessed by Western blotting.
Little or No Binding of vMIP-II-Derived Peptides to CXCR7
We used antibody receptor competition assays to further evaluate the weak inhibitory effect of DV1 on Akt phosphorylation in CXCR7-transfected CHO cells and to confirm the selectivity of vMIP-II-derived peptides for CXCR4. Both peptides, but especially HC4319 (Fig. 3), showed only very slight binding to CXCR7. This lack of binding may explain why HC4319 and DV1 had little or no effect on the phosphorylation of Akt in CXCR7-transfected CHO cells.

Lack of binding of HC4319 and DV1 to CXCR7. The binding of HC4319 and DV1 in CXCR7- or CXCR4-transfected CHO cells was detected by antibody competition binding assays. The results shown are the mean values of at least three independent experiments.
Significant Mobilization of HPCs to the Blood by vMIP-II-Derived Peptides in C3H/HeJ mice
We used an HPC assay to investigate the effect of vMIP-II-derived peptides on HPC mobilization in C3H/HeJ mice. Administration of either the monovalent peptide (37.5 and 150 mg/kg) or the bivalent peptide (57.5 and 230 mg/kg) caused a rapid and significant mobilization of HPCs to the peripheral blood 1 h after subcutaneous administration to C3H/HeJ mice. The numbers of BFU-Es, CFU-GMs, and CFU-GEMMs in the peripheral blood were ∼5–10 times greater than those in the control group (Fig. 4).
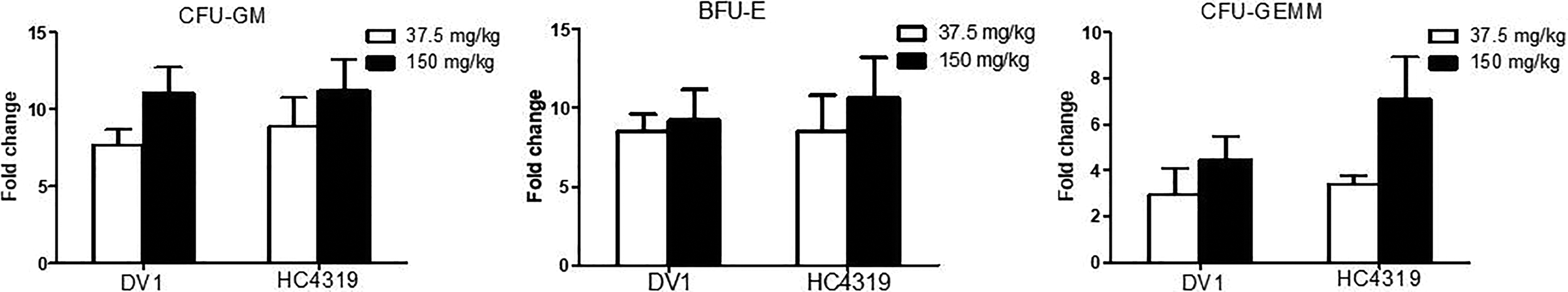
Significant mobilization of HPCs to the blood in C3H/HeJ mice by HC4319 and DV1. Both HC4319 and DV1 caused strong mobilization of HPCs from the bone marrow to the peripheral blood 1 h after subcutaneous administration compared with the control group (P < 0.01) administered vehicle only. The results shown are the mean values of at least three independent experiments.
Discussion
Previous studies by many groups, including ours, have shown that the N-terminus of vMIP-II is the critical determinant for CXCR4 recognition 20,25 –30 . The synthetic peptides DV1 and DV3, which contain 21 and 10 D-amino acids, respectively, from the N-terminus of vMIP-II (Table 1), displayed selective binding to CXCR4 with half-maximal inhibitory concentration (IC50) values of 236 and 440 nM, respectively. Mutational studies of DV1 peptide indicated that N-terminal residues 1–10 were essential for CXCR4 binding, whereas C-terminal residues 11–21 played a secondary role in binding to CXCR4 21 . We generated HC4319 by chemically linking DV1 and DV3, a shortened version of DV1 that contains the essential first 10 residues of DV1 as described above, via the side chain of a D-Lys residue. Previous binding studies using the antibody receptor competition assay on HEK293 cells that stably expressed CXCR4 demonstrated that HC4319 has very high affinity for CXCR4, with an IC50 value of ∼4 nM, and is much more potent than the monomeric counterparts DV1 and DV3 (Table 1). In the chemotaxis assays using SUP-T1 cells, HC4319 could block the cell migration induced by SDF-1a and the IC50 value was 500 nM 22 .
Sequences and IC50 Values of CXCR4 Competitive Binding of DV-1, HC4319, and vMIP-II.

a Underlined text indicates D-amino acids.
In this study, we found that two of the above described peptides derived from the N-terminus of vMIP-II, HC4319 and DV1, inhibited the phosphorylation of Akt and Erk induced by SDF-1α significantly. Because CXCR7 (RDC1) is the second identified receptor of SDF-1α and its affinity for SDF-1α is higher than that of CXCR4 for SDF-1α 23,24 , we also examined the effects of SDF-1α on the phosphorylation of Akt and Erk in CXCR7-transfected CHO cells or wild-type CHO cells. Our data showed that only weak phosphorylation of Akt was induced by SDF-1α in CXCR7-transfected CHO cells and that vMIP-II-derived peptides had little inhibitory effect on Akt phosphorylation in CXCR7-transfected CHO cells, which was consistent with their lack of binding to CXCR7.
Many novel synthetic peptides derived from the N-terminus of vMIP-II, including bivalent and D-peptides, were reported previously to have significant antiviral activity by inhibiting the replication of CXCR4-dependent HIV-1 strains 21,28,30 . However, the in vivo activity of these peptide inhibitors of CXCR4 as HSC mobilizers in animal models has not been studied. This is the first report that these bivalent and D-peptides have a strong in vivo HPC mobilization effect. In general, D-peptides containing unnatural D-amino acids are thought to be more resistant to enzymatic degradation than natural L-peptides, which may result in higher stability and be advantageous for in vivo and eventual clinical applications. For example, a D-peptide agonist of the calcium-sensing receptor AMG 416 (velcalcetide) has been approved recently for the treatment of secondary hyperparathyroidism in hemodialysis patients 31 –33 . Therefore, the in vitro and in vivo activities of our synthetic bivalent and D-peptide inhibitors of CXCR4 reported here suggest that these novel molecules merit further development as new therapeutic agents for mobilizing HSCs to the peripheral blood in regenerative medicine.
Footnotes
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
All animal experiments were approved by the Laboratory Animal Research Center of Tsinghua University.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from Tsinghua University and the Tsinghua-Peking Joint Center for Life Sciences (to Z. H.), the National Institutes of Health (GM 57761, to Z.H and J.A.), and the California Institute for Regenerative Medicine (RS1-00225 -1, to Z.H.).