
Abstract
Attaining consistent robust engraftment in the structurally normal liver is an obstacle for cellular transplantation. Most experimental approaches to increase transplanted cells’ engraftment involve recipient-centered deleterious methods such as partial hepatectomy or irradiation which may be unsuitable in the clinic. Here, we present a cell-based strategy that increases engraftment into the structurally normal liver using a combination of magnetic targeting and proliferative endoderm progenitor (EPs) cells. Magnetic labeling has little effect on cell viability and differentiation, but in the presence of magnetic targeting, it increases the initial dwell time of transplanted EPs into the undamaged liver parenchyma. Consequently, greater cell retention in the liver is observed concomitantly with fewer transplanted cells in the lungs. These highly proliferative cells then significantly increase their biomass over time in the liver parenchyma, approaching nearly 4% of total liver cells 30 d after transplant. Therefore, the cell-based mechanisms of increased initial dwell time through magnetic targeting combined with high rate of proliferation in situ yield significant engraftment in the undamaged liver.
Introduction
Cell transplantation may be a viable alternative to orthotopic liver transplantation for the treatment of liver-based inborn errors of metabolism. 1,2 However, there are few reports (preclinical or otherwise) of long-term engraftment and repopulation of the recipient organ with transplanted cells without preconditioning liver injury. In patients severely affected by urea cycle defects, such as neonatal onset of Ornithine transcarbamylase (OTC) deficiency, hepatocyte transplantation can provide a short-term therapeutic effect, but recipients require either repeated doses of transplanted hepatocytes or ultimately liver transplant 3,4 presumably due to the low proliferative capacity and high turnover rate of transplanted hepatocytes. Thus, hepatocyte transplantation can be an effective strategy to delay organ transplant in urea cycle defect patients but fails to provide a long-term solution.
Experimental methods to increase liver engraftment efficiency of a variety of transplanted cell types in rodent models require extensive parenchymal injury to the recipient liver, 5 –7 and these methods are not easily translated to the clinical setting. 8 Clinical methods of increasing engraftment include irradiation or portal vein embolization, 8,9 which still involve risk to the recipient including morbidity. 10 –12 Therefore, a cell transplantation–based strategy that can be used in the clinic (i.e., does not require liver injury in the recipient) and results in long-term persistence of transplanted cells would increase patient quality of life and reduce the demand for liver transplant.
Transplantation of partially differentiated stem cells may provide a long-term solution for repopulation of the quiescent liver independent of liver damage. We have shown that endoderm progenitor (EP) cells, which are early derivatives of embryonic stem cells, engraft into the quiescent liver without preconditioning damage and reverse the phenotype of a mouse model of hemophilia B. 13 Although effective, the observed engraftment was quantitatively inconsistent, leading us to test both in vitro and in vivo strategies to standardize engraftment and increase its efficiency. In this study, we find that using a more proliferative EP cell, combined with magnetic targeting to the liver, increases the efficiency and reproducibility of engraftment in the quiescent liver independent of preconditioning damage. These findings together with our previous studies 13 suggest the use of EP cells and magnetic targeting may provide a long-term strategy to reduce disease phenotype of liver-based inborn errors of metabolism.
Materials and Methods
Mouse ES Cell Culture and Differentiation
The ES cells used are derived from strain 129P2_Ola mice and constitutively expresses green fluorescent protein (GFP) driven by the β-actin promoter. 13,14 Cells were maintained on murine embryonic fibroblast feeder layers that produce leukemia inhibitory factor. Embryonic stem cell (ESC) propagation media was high glucose Dulbecco’s modified Eagle’s medium (DMEM) (Sigma, USA) supplemented with 15% Fetal Bovine Serum (FBS) (Sigma), 0.1 mM 2-mercaptoethanol (Sigma), 100 units/mL penicillin, and 100 g/mL streptomycin (GIBCO, USA). 13 To ensure viability and pluripotency, ESC media were changed daily, and cells were passed every 2 to 3 d. Cells were passed using trypsin (0.05% trypsin, in 0.53 mM ethylenediaminetetraacetic acid [EDTA]) for 3 min followed by filtration through a 40-µm strainer to remove differentiated cells. After at least 2 passages from frozen stocks, the cells were used for experiments.
Two different methods were used for endoderm induction: (1) mouse ES cells were plated at 4 × 105 cells/collagen-coated 35 mm2 dish and grown in ESC propagation media supplemented with 100 ng/mL acidic fibroblast growth factor (FGF) (Sigma) for 5 to 7 d as described 13 or (2) embryoid bodies were collected after 2.5 d then subjected to a 2-step protocol using serum-replacement media supplemented with 100 ng/mL human activin A (R&D Systems, USA) as described. 15 Trypan blue exclusion for cell viability was performed as described, 16 and growth curves were assessed by trypan blue excluding cells counted in both technical and biological triplicate during the differentiation time course and used to calculate doubling times. The BrdU/7AAD staining assay was performed as described by the manufacturer (BD Pharmingen, USA). Hepatic differentiation was induced using day 8 endoderm cells (from acidic FGF method, see above) incubated in Hepatocyte Culture Media (HCM, Lonza, USA) supplemented with 30 ng/mL FGF4 (R&D Systems, Minneapolis, USA) and 20 ng/mL bone morphogenetic protein 2 (BMP2) (R&D Systems) for 2 d, followed by 4 d in HCM supplemented with 20 ng/mL hepatic growth factor (HGF) (R&D Systems), 10 ng/mL oncostatin M (R&D Systems), and 0.1 µM dexamethasone (R&D Systems).
Superparamagnetic Microsphere (SPM) Labeling of EP Cells
EPs were labeled with fluorescent (flash red) SPM particles (0.9 µm diameter; Bangs Laboratories, Fishers, IN, USA) by incubation in cell culture for 24 h. Successful labeling was confirmed by flash-red fluorescence. Labeling efficiency was assessed by flow cytometry. In vitro toxicity experiments were performed 24 h after SPM labeling. Cell viability was assessed by trypan blue exclusion. Apoptosis and necrosis were assessed by flow cytometry using 7AAD and annexin-V stains (BD Pharmingen, San Jose, CA, USA). Albumin (Alb) and α-fetoprotein messenger RNA (mRNA) abundance were measured by quantitative real-time polymerase chaing reaction (PCR) (see below).
Animals
Wild-type Balb/c mice were obtained from Jackson Laboratory and housed in the Animal Care Services Facility at the University of Florida. All mice weighed from 15 to 25 g. Mice were maintained on standard chow and kept on a 12-h light/dark cycle. All procedures performed were approved by the Institutional Animal Care and Usage Committee at their respective institutions and thus in compliance with the guidelines for humane care of laboratory animals.
Transplantation of EP Cells
EPs (1 × 106) were suspended in phosphate buffered saline (PBS) with 1% fetal bovine serum (FBS) and 0.1 mM ethylenediaminetetraacetic acid (EDTA) and incubated on ice for no more than 30 min while awaiting transplant. Mice were anesthetized with an intraperitoneal (IP) injection of ketamine and dexmedetomidine or inhaled isoflurane. A small incision was created either below the right costal margin or along the midline. The cell suspension was injected into the portal vein using a 30-G needle with a magnet in place on the adjacent body wall. Atipamezole (1.0 mg/kg, IP) was used to reverse anesthesia if ketamine were used. Mice were dosed with 0.1 g/kg IP buprenorphine after surgery and monitored closely thereafter. Following skin wound closure, a 1.3 Tesla magnet (K&J Magnetics, Inc.) sheet (15 × 15 × 2 mm3) was attached to the skin with a bandage at the right lower rib cage and was kept in place for 18 h.
Hepatocyte Isolation and Fluorescence Activated Sorting of GFP Positive Cells
Hepatocytes were isolated from mouse livers after EP + SPM injection by a sequential perfusion method as previously described. 17 Briefly, the inferior vena cava was cannulated with an 18 G intravenous (IV) catheter. The portal vein was severed and the suprahepatic vena cava was tied off. The liver was then perfused first with an EGTA solution, followed by collagenase at 100 collagen digestion units (CDU/mL). The resulting cell suspension had greater than 80% viability as measured by the trypan blue exclusion assay and was then analyzed and sorted using a FACSAria II flow cytometer (BD Biosciences) equipped with a combination of 407, 488, 561, and 640 nm lasers. Sorted cells were deflected into Falcon tubes containing PBS + 20% fetal calf serum.
Quantitative RT-PCR
Real-time fluorescent quantitative PCR was performed as described previously. 13 Briefly, total RNA was harvested and purified using the RNeasy Mini Kit (Qiagen). One microgram was reverse transcribed using the QuantiTect Reverse Transcription Kit (Qiagen) following the manufacturer’s directions. The resultant cDNA template was diluted 50-fold and 100 pg of template amplified in RT2 SYBR Green/ROX qPCR Master Mix (SABiosciences-Qiagen) on an ABI 7300 optical thermocycler (Applied Biosystems). For each amplification, a no template control was performed to ensure no detectable product was formed from spurious primer-only amplification. Relative levels of mRNA were normalized to β-actin, which was used as the internal control. The primer sequences were used for β-actin (forward: 5′-ATGCTCCCCGGGCTGTAT-3′, reverse: 5′-CATAGGAGTCCTTCTGACCCATTC-3′), α-fetaprotein (Afp; forward: 5′-ATTGCCTCCACGTGCTGCCA-3′, reverse: 5′-GAAAATGTCGGCCATTCCCT-3′), Alb (forward: 5′-GGCACCAAGTGTTGTACACT-3′, reverse: 5′-AGCAGACACACACGGTTCAG-3′), hepatocyte nuclear factor 4 alpha (Hnf4a; forward: 5′-ACACGTCCCCATCTG AAG-3′, reverse: 5′-CTTCCTTCTTCATGCCAG-3′), Cyp3a (forward: 5′-TGGGTGAGTGGT TGCTTACA-3′, reverse: 5′-GAGGGAAACTGGTGAGGATG-3′), transferrin (forward: 5′-GGTCCCTCGAAAGATAGACATCA-3′, reverse: 5′-GGGAGTCTTCCAGACCTC TTTTAA-3′), and α-1-antitrypsin (a-1-AT; forward: 5′-AATGGAAGAAGCCATTCGAT-3′, reverse: 5′-AAGACTGTAGCTGCTGCAGC-3′).
3-D Microscopy
Fresh liver explants were examined with a stereomicroscope (MZ16FA, Leica Microsystems) using a GFP2 long-pass filter (100447084, Leica Microsystems) to detect the presence of GFP-positive cells.
Statistical Analysis
Results are presented as mean ± standard deviation from 3 independent biological replicates unless specified otherwise. Comparisons between 2 groups were performed using 2-tailed unpaired Student’s t-test, and comparisons consisting of more than 2 groups were performed using two-way analysis of variance. Differences were considered statistically significant when P ≤ 0.05 and are noted as such where applicable.
Results
Cell Proliferation Rate Correlates with Engraftment in Quiescent Liver
Initially, the aim of this study was to compare different endoderm differentiation methods for differentiation efficiency, cell proliferation, and viability rates and correlate these with engraftment efficiency in undamaged mouse liver. We hypothesized a more efficiently differentiated EP cell population that was highly proliferative and viable would engraft more readily in the quiescent liver. We previously measured markers of endoderm (Sox17, FoxA2, and Gata4), mesoderm (Nkx2.5, goosecoid), ectoderm (nestin, Pax6), pluripotent (Oct4), and hepatic (Afp, Alb) gene expression in acidic fibroblast growth factor (aFGF) differentiation time courses and find efficient induction of endoderm transcripts and proteins, but low to undetectable levels of other lineage marker mRNAs. 13,14,18,19 Comparing these results to those obtained using the ActivinA differentiation method 15 indicated induction of various endoderm marker mRNAs and that pluripotency-related transcripts are also reduced using each differentiation protocol. 15,18,19 Additionally, we detected very few dead cells during both the aFGF and ActivinA 6-d differentiation time course (Fig. 1A and data not shown), indicating no significant difference in cell viability between the 2 methods. Therefore, we conclude these 2 differentiation methods yield efficiently differentiated EP cell populations with a low level of cell death.

High proliferation rate positively correlates with endoderm progenitor (EP) cell liver engraftment. (A) Trypan blue exclusion assay was performed on spontaneously differentiated ES cells or ES cells undergoing the aFGF or Activin A methods for 6 d to generate growth curves. Average cell numbers for each day were recorded from biological triplicate cultures (error bars represent standard deviation [SD] from the mean) and used to calculate doubling time for each culture condition. (B) BrdU/7AAD staining was performed on day 7 differentiated aFGF-EPs and Activin–EPs and analyzed by flow cytometry to determine cell cycle phase distribution of biological triplicate cultures with error bars representing SD from the mean. (C) Representative image of whole liver analyzed by stereomicroscopy using fluorescein isothiocyanate (FITC) filter to identify green fluorescent protein-positive cells 14 d after aFGF-EP transplant (10× magnification).
In contrast, we observe a striking difference in the proliferation rate of EPs produced from these 2 different endoderm differentiation protocols: EP cells produced from the aFGF (aFGF-EPs) method have a significantly higher proliferation rate (doubling time of 19.5 h) compared to cells from the ActivinA method (activin-EPs) with doubling time of 28.7 h (Fig. 1A; P ≤ 0.01). A complementary approach supports this finding, as a significantly greater percentage of aFGF-EP cells are in S phase of the cell cycle (Fig. 1B; P ≤ 0.01) as determined by BrdU/7AAD staining and flow cytometry analysis. Therefore, aFGF-EPs and activin-EPs have similar endoderm and pluripotency marker gene expression profiles and levels of cell viability, but aFGF-EPs proliferate at a significantly higher rate. We next tested the liver engraftment efficiency of EPs by portal vein injection in Balb/c mice and analysis of whole liver explant using fluorescent stereomicroscopy, 20 which allows us to detect GFP+ cells several millimeters deep within the organ (see online Fig. S1 for experimental overview). Fourteen days after transplant of aFGF-EPs and activin-EPs, we readily detected transplanted GFP-positive aFGF-EP cells in liver explants (Fig. 1C and consistent with our previous observations 13 ) but were unable to detect GFP-positive activin-EP cells under the same conditions (N = 3 each). These findings support the conclusion that a more proliferative EP cell may be a superior engraftment candidate for delivery to the undamaged liver parenchyma.
SPM-labeled EP Cells Maintain Viability and In Vitro Differentiation Capacity
Based on the above results, we focused on using the aFGF-EP and reasoned that enhancing early transplant events such as cell delivery and initial dwell time in the liver (independent of preconditioning injury) would further contribute to engraftment efficiency of aFGF-EPs. Magnetic targeting using cells labeled with superparamagnetic (SPM) nanoparticles increases engraftment into disparate target tissues, 21 –24 therefore this approach may further increase engraftment of aFGF-EPs in the undamaged liver. This process involves the endocytosis of iron particles (SPMs) by cells of interest, which can then be attracted and retained by a magnetic field. 25 Because of iron’s potential toxicity, we first tested whether internalization of SPMs affected cell viability or differentiation.
aFGF-EPs were differentiated for 6 d then incubated with SPMs conjugated to flash-red fluorophore at a ratio of 500:1; 1,000:1; and 2,000:1 (SPM:cell) for 24 h to determine optimal dosage for cell labeling and potential toxicity of SPM incorporation into EP cells. We confirmed particle uptake by aFGF-EPs that constitutively express GFP using both confocal microscopy (Fig. 2A) and flow cytometry (Fig. 2B). The average percentage of SPM-labeled cells after 24 h is 83.6% ± 3.1 (Fig. 2C), indicating label saturation at a ratio of 500:1, as no increase in cell labeling is detectable at higher ratios (Fig. 2C). Using the trypan blue exclusion assay, we observe no significant change in cell viability at the 500:1 (SPM:cell) ratio; however, both the 1,000:1 and 2,000:1 ratios are correlated with significant increases in cell death (Fig. 2D; P ≤ 0.05 and P ≤ 0.01, respectively). However, measurement using the more sensitive annexin-V/7-AAD assay (Fig. 2E) indicates a statistically significant, but nominal, increase in apoptotic and necrotic cells of 1% each using the 500:1 ratio (Fig. 2F). Therefore, a ratio of 500:1 efficiently labels aFGF-EPs without adversely affecting cell viability and causes them to be attracted and retained by a magnetic field in vitro (online Fig. S2).
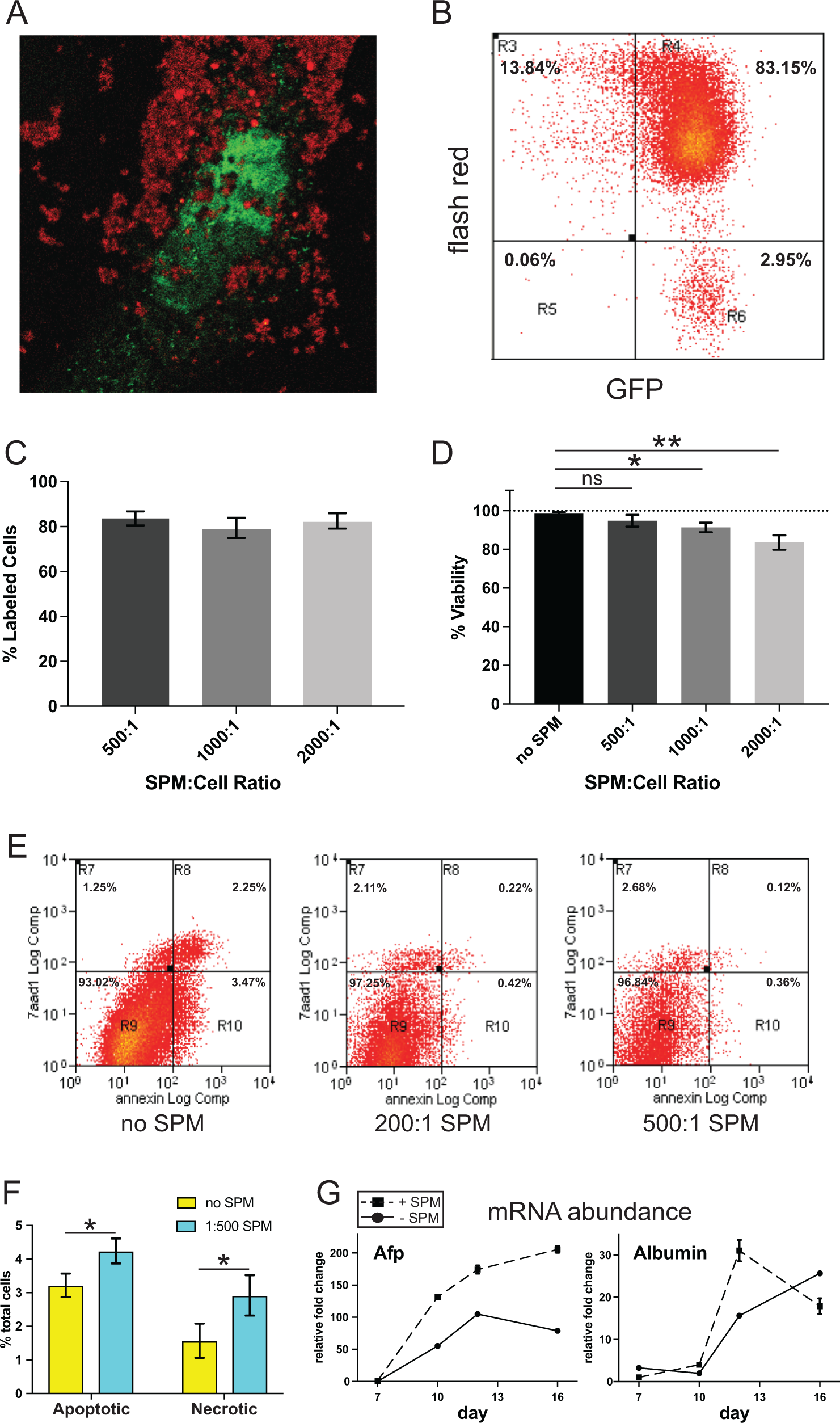
Superparamagnetic microsphere (SPM) labeling does not deleteriously affect aFGF-endoderm progenitor (EP) viability or hepatic lineage progression. (A) Day 7 differentiated aFGF-EP cells were incubated with flash-red fluorescent chromosphere labeled SPMs for 24 h and analyzed by confocal microscopy at 60× magnification; image shows a single cell with nuclearlocalized green fluorescent protein. (B) aFGF-EP cells were labeled with SPMs as in (A) and analyzed by flow cytometry. (C) Day 7 differentiated aFGF-EP cells were incubated with
To test if SPM labeling disrupts the differentiation process, day 6 aFGF-EPs were labeled with SPMs as above, then differentiated toward the hepatic lineage, and markers for hepatoblast (Afp) and hepatocyte (Alb) formation were measured 1, 3, 5, and 10 d post-SPM labeling. Measurement of Afp and Alb mRNAs shows comparable induction over the differentiation time course between cells incubated with and without SPM (Fig. 2G). This indicates the in vitro differentiation potential of these cells to form hepatoblast- and hepatocyte-like cells is not compromised by SPM labeling. In summary, SPM labeling aFGF-EPs neither drastically alters cell viability nor interrupts the hepatic differentiation program in vitro; therefore, SPMs may be a suitable tool to boost in vivo liver engraftment independent of liver injury.
Magnetic Targeting Increases aFGF-EP Engraftment Efficiency in Undamaged Liver
SPM labeling aFGF-EP cells followed by magnetic targeting may be an effective method to increase the initial dwell time of transplanted cells to recipient undamaged liver parenchyma. Accordingly, we hypothesize an increase in dwell time combined with a proliferative transplanted cell may be sufficient to achieve quantitatively significant engraftment independent of preconditioning liver damage. To test if magnetic targeting can increase liver engraftment of FGF-EPs in vivo, we injected SPM-labeled GFP-positive aFGF-EPs into the portal vein of wild-type Balb/c mice with or without a 1.3 Tesla magnet applied to the outer body wall during and 18 h postinjection and monitored engraftment efficiency. Initial engraftment rates drop dramatically after 48 h, 26 so the presence of GFP-positive cells from collagenase-digested recipient livers was assessed at multiple time points: 30 min, then 1, 3, 7, and 30 d posttransplant by flow cytometry. Consistent with enhancement of the initial dwell time in the liver, we observe a significant increase in percentage of GFP-positive cells 30 min posttransplant using magnetic targeting (Fig. 3A). In fact, at each time point analyzed, there is a significantly higher percentage of GFP-positive cells when using magnetic targeting enhanced aFGF-EP transplant compared to control transplants without magnetic targeting (Fig. 3A). Consistent with previous results, 26 we also observe an initial and steady decline in GFP-positive cell numbers in situ (30 min to 3 d); however, the significant increase between 7 and 30 d (Fig. 3A) suggests a period of exponential cell growth in vivo. Additionally, qualitative analysis of engraftment in fresh frozen tissue sections of livers at 30 d posttransplant shows readily detectable fluorescent clusters of cells, similar to previous observation, 13 but more robust clusters are detected with magnetic targeting (Fig. 3B). We conclude that magnetic targeting significantly increases aFGF-EP engraftment efficiency in the undamaged liver, in part by increasing the initial dwell time within the parenchyma.

Magnetic targeting increases liver engraftment efficiency of aFGF-endoderm progenitors (EPs). (A) Day 7 differentiated green fluorescent protein (GFP)+ aFGF-EPs were incubated with superparamagnetic microspheres (SPMs) at a 1:500 ratio for 24 h and injected into mouse portal vein with (+magnet) or without (−magnet) magnets fixed on the outer body wall. Engraftment was assessed at 0.5 h, 1, 3, 7, and 30 d after transplantation by identification of GFP+ cells from livers perfused to a single-cell suspension determined by flow cytometry
To determine whether magnetic targeting has any adverse effect on terminal differentiation of aFGF-EPs in vivo, GFP-positive cells were retrieved from mouse livers 30 d posttransplant by fluorescence activated cell sorting (FACS) (Fig. 3C), and mRNA abundance was measured for liver-specific mRNA expression (Fig. 3D). We compared mRNA abundance from transplanted GFP+ cells retrieved from the liver after 30 d, along with native mouse hepatocytes (positive control), day 7 differentiated aFGF-EPs (undifferentiated control pretransplant), and spleen (negative control) by RT-qPCR for the liver-specific mRNAs Alb, HNF4A, Cyp3a, transferrin, and α1 antitrypsin. The results indicate the engrafted GFP+ cells have a very similar gene expression profile to hepatocytes (Fig. 3D), thus SPM-labeled EPs engraft then differentiate to hepatocyte-like cells in vivo.
Since magnetic targeting appears to promote retention of aFGF-EPs in the liver parenchyma immediately and up to 1 wk after transplant (Fig. 3A), we hypothesized this strategy might inhibit vascular flow-through of transplanted cells into the lungs. If so, the transplanted cells would indeed have enhanced dwell time within the liver, which would aid engraftment within the parenchyma and subsequent proliferation in situ. A potential unintended consequence of this, though, is magnetic field-enhanced dwell time/retention could lead to cell clumping resulting elevated stasis and subsequent thrombosis, as has been reported in hepatocyte transplantation. 27 To address this possibility, we examined the transplanted cells’ localization patterns within the sinusoid of livers very early during the engraftment process, as cell clumping/cohesion will be readily apparent if it is a major consequence of magnetic targeting. Stereomicroscopy of liver explant 30 min after delivery of SPM-EPs shows GFP+ cells distributed diffusely and stochastically throughout the sinusoid. However, 24 h posttransplant a characteristic pattern of localization around the periphery of the sinusoid is evident for aFGF-EPs, independent of magnetic targeting (online Fig. S3; N = 3 each), which is consistent with localization patterns observed at later time points (online Fig. S1 [4 d] and also see Fig. 3E [7 d]), suggesting that occlusion of the portal vessels does not contribute to magnetic targeting-based enhancement of engraftment. Furthermore, we determined the number of GFP+ cells per area of microscopic field of view (Fig. 3E) in both the liver and the lungs of mice receiving transplanted aFGF-EPs with or without magnetic targeting 7 d posttransplant. Delivery enhanced with magnetic targeting results in significantly fewer GFP+ cells in the lung (P ≤ 0.05) and more GFP+ cells in the liver (P ≤ 0.01) per square millimeter area than without magnetic targeting (Fig. 3F). Taken together, these results indicate magnetic targeting aFGF-EPs increases overall dwell time of aFGF-EP cells in the liver, significantly increasing the quantitative engraftment of these cells in the undamaged liver parenchyma without the development of portal occlusion and thrombosis.
Discussion
We present here a strategy that can be rapidly adopted in the clinic to increase the initial engraftment of transplanted cells into the quiescent, undamaged liver parenchyma. Previous studies utilizing hepatocyte transplantation have required parenchymal damage, that is, hepatectomy or retrorosine, for significant engraftment to occur. 7,8,28,29 Even then, multiple injections may be necessary to attain a therapeutic effect. 26,30 Justifying these strategies for patients with metabolic alterations but otherwise normal livers may be difficult; indeed, hepatocyte transplantation in OTC deficient patients only provides a short-term benefit while awaiting liver transplant. 3,4,31 Furthermore, the relatively large size of hepatocytes at ∼40 μm and a report of portal thrombosis in a hepatocyte transplant recipient 27 presents additional challenges. Our cell transplant strategy combining magnetic targeting with proliferative, relatively small EPs (∼20 μm 18,19 ) provides a novel proof-of-concept that long-term, quantitatively significant engraftment can be achieved in the undamaged mouse liver. These findings along with our previous study indicate aFGF-EP cells can differentiate and function as hepatocytes in vivo, as they are able to produce factor IX and reverse a hemophilic phenotype. 13
The striking difference in proliferation between the aFGF-EP and activin-EP cells derived from identical mouse ES cell cultures positively correlates with liver engraftment 2 wk posttransplant (Fig. 1). It is likely that delivery of these cells via portal vein injection is relatively inefficient and that Kupffer cells may clear many transplanted cells, 30 but a proliferative cell that persists in the undamaged liver parenchyma is able to undergo more doublings than a less proliferative cell. This is supported by the observed increase in GFP+ cells retrieved from livers comparing 7 to 30 d posttransplant (Fig. 3A). Although we observed no teratoma or other tumor formation in the current study, a highly proliferative cell will need to be more fully vetted as unchecked proliferative activity can lead to DNA damage and tumorigenesis. 32 Therefore, future experiments will examine this possibility using human stem cell derivatives transplanted into quiescent livers of rodent models.
Magnetic targeting to enhance aFGF-EP engraftment into the undamaged liver parenchyma has several advantages. First, magnetic targeting results in a higher proportion of transplanted cells in the liver immediately after delivery and up to 30 d postdelivery (Fig. 3A), indicating this strategy both increases the initial dwell time and the biomass ultimately generated by transplanted aFGF-EPs after 30 d. Second, based on a focused panel of gene expression measurements, magnetic targeting does not appear to negatively affect differentiation of aFGF-EPs in vitro (Fig. 2G) or in vivo (Fig. 3D), suggesting these hepatocyte-like cells could reverse a liver-based disease as we have previously demonstrated. 14 Additionally, transplanted aFGF-EP cells may partially differentiate in vivo and form a population of liver stem cells capable of self-renewal and differentiation; however, further study is required to address this question. Third, we observe a significantly higher retention of transplanted cells in the liver versus the lung (Fig. 3E), suggesting a reduction in cellular pulmonary emboli that may be clinically significant. Also, since aFGF-EPs are much smaller than hepatocytes (cf. ∼20 to ∼40 μm), there is less risk of portal embolism. We do not observe overt evidence of portal stasis or inflammation in the current study either with or without magnetic targeting; however, additional studies are required to address these possibilities as well as determine how transplanted cells pass through the portal vasculature and presumably migrate through the space of Disse to ultimately colocalize with native hepatocytes. The sequence of events that results in long-term engraftment of EPs in the hepatocyte space is likely different from other cell types tested thus far but needs further delineation.
In conclusion, this approach represents an easily scalable, novel, and promising proof of concept that has the potential to translate directly to therapeutic strategies for specific liver-based genetic disorders.
Footnotes
Authors’ Note
W. Samuel Fagg and Naiyou Liu contributed equally to this work.
Ethical Approval
This study was approved by our Institutional Animal Care and Use Committee (IACUC).
Statement of Human and Animal Rights
Animal procedures were approved by IACUC and monitored by institutional veterinary staff.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by US National Institutes of Health, grant number HL082606-0 (Jeffrey Fair); the NIH National Institute of Diabetes and Digestive and Kidney Diseases, grant numbers DK079879 and DK090115 (Jae-Sung Kim); National Institute on Aging, grant number AG028740 (Jae-Sung Kim); and CIRM, grant number TG2-01157 (W. Samuel Fagg).
Supplemental Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.