
Abstract
Despite having pathological changes in the brain associated with Alzheimer's disease (AD), some patients have preserved cognitive function. A recent epidemiological study has shown that diet, exercise, cognitive training, and vascular risk monitoring interventions may reduce cognitive decline in at-risk elderly people in the general population. However, the details of molecular mechanisms underlying this cognitive function preservation are still unknown. Previous reports have demonstrated that enriched environments prevent the impairment of hippocampal long-term potentiation (LTP) through β2-adrenergic signals, when LTP is incompletely suppressed by synthetic amyloid-β (Aβ) oligomers. The cholinergic network from the medial septal nucleus (MSN) is also a main modulating system for hippocampal glutamatergic neural activation through nicotinergic and/or muscarinergic acetylcholine receptors. Previously, we reported the importance of a cholinergic regulator gene in the MSN, hippocampal cholinergic neurostimulating peptide (HCNP). By using hippocampal sections from mice, we here demonstrated that the cholinergic neural activation from the MSN enhanced the glutamatergic neuronal activity during unsaturated LTP but not during saturated LTP. Synthetic Aβ oligomers suppressed the hippocampal glutamatergic activity in a concentration-dependent manner. Furthermore, HCNP, as well as a cholinergic agonist acting through the muscarinic M1 receptor, prevented the suppression of hippocampal glutamatergic neuronal activity induced by synthetic Aβ oligomers. This result suggests that the persisting cholinergic activation might be a potential explanation for the individual differences in cognitive effects of AD pathological changes.
Keywords
Introduction
Senile plaques, neurofibrillary tangles, and neuronal cell death are the neuropathological hallmarks of Alzheimer's disease (AD). 1 Amyloid-β (Aβ) is the main pathogenic feature of AD and forms the basis for the amyloid hypothesis of AD. 2 However, several therapeutic interventions reducing Aβ have failed to ameliorate the loss of cognitive function in patients with AD. Conversely, there is evidence that some people can tolerate more AD-related pathological changes than others, having “cognitive reserve.” 3,4 In individuals with cognitive reserve, the functional network connection in the cerebral cortex can be visualized as intact using functional magnetic resonance imaging (fMRI). 5 Interestingly, recent studies have suggested that a nutritious diet, physical activity, social engagement, and mental stimulation inhibit cognitive decline in aged individuals. 6 –10 An intervention study of diet, exercise, cognitive training, and vascular risk monitoring also revealed a reduction in the decline of cognitive function in at-risk elderly people in the general population. 11 This epidemiological evidence indicates the possibility of maintaining cognitive function even if AD-related pathological changes, such as amyloid deposition and aggregation of tau protein, occur within the brain. However, the details of the molecular mechanisms underlying the preservation of cognitive function in the presence of AD-related pathological changes are still unknown.
Much AD research has focused on the septal cholinergic neurons because they degenerate early in the disease progression. This observation is validated by the clinical usefulness of cholinesterase inhibitors. Hippocampal function is dynamically regulated by postsynaptic transmission in the mono- and tri-glutamatergic pathways from the dentate nucleus to CA1 area. The cholinergic neural formation from the medial septal nuclei (MSNs) to the hippocampus regulates the hippocampal glutamatergic neural activity. 12 Cholinergic dysfunction is involved in memory disturbances in AD and schizophrenia. 13 This suggests that the septo-hippocampal cholinergic network may be of crucial importance for the regulation and maintenance of glutamatergic neural activity in the hippocampus.
Hippocampal cholinergic neurostimulating peptide (HCNP), originally purified from juvenile rat hippocampus, was first reported as an acetylcholine synthesis regulating peptide in the MSN by Ojika et al. in 1992. 14,15 HCNP induces acetylcholine synthesis by increasing the amount of choline acetyltransferase in the MSN. This peptide is formed at the N-terminal region of the 21-kD HCNP precursor protein (HCNP-pp), which is composed of 186 amino acids. We have also reported that overexpression of HCNP-pp in the hippocampus may enhance the glutamatergic neuronal activity, measured by the slope of field excitatory postsynaptic potentials (fEPSPs) during long-term potentiation (LTP), via the muscarinic M1 receptor, indicating that it is a factor regulating the hippocampal glutamatergic neurons. 16 A previous report also demonstrated that synthetic amyloid oligomers generated by mutant Aβ 1 to 40 peptides (AβS26C, serine to cysteine at amino acid 26) could inhibit glutamatergic neuronal activity in a concentration-dependent manner. 17
In this study, we investigated whether overexpression of HCNP can enhance LTP in the hippocampus when synthetic amyloid oligomers inhibit glutamatergic neural activation. We report that cholinergic neural activity enhances glutamatergic neuronal activity, as measured by the fEPSP slope in unsaturated, but not saturated, LTP. Furthermore, overexpression of HCNP (HCNP-pp), as well as the cholinergic agonist, carbachol (CCh), rescued the glutamatergic neural activity from reduction by synthetic amyloid oligomers.
Materials and Methods
Animals
Male 6- to 8-wk-old C57BL/6J mice were used in experiment 1. And 12- to 15-wk-old HCNP-pp heterozygous transgenic (HCNP-pp Tg) mice, driven by a Ca2+ CaMKII promoter, and their wild-type (WT) littermates were used in experiment 2. All animals were kept under pathogen-free conditions on a 12 h light/dark schedule (light on 07:00 to 19:00) and given free access to food and water. These experiments were approved by the Animal Care and Use Committee of Nagoya City University Graduate School of Medical Sciences and conformed to guidelines for the use of laboratory animal published by the Japanese government (Law No. 105, October 1973).
Slice Preparation
Mice were deeply anesthetized with halothane and decapitated. The brains were quickly removed and transverse hippocampal slices with a 400-µm thickness were prepared using a vibrating slice cutter (Linear Slicer Pro 7, Dosaka, Kyoto, Japan) in ice-cold solution containing (millimole: mM) sucrose, 260; KCl, 3; NaH2PO4, 1.25; NaHCO3, 26; D-glucose, 10; MgCl2, 1 (pH 7.4), which was continuously bubbled with 95% O2/5% CO2. One to 4 slices were obtained from each animal and 1 experiment was performed on each slice. The hippocampal slices were incubated for 30 min in artificial cerebrospinal fluid (ACSF) containing (mM) NaCl, 125; KCl, 2.5; CaCl2, 2.4; MgCl2, 1; NaH2PO4, 1.25; NaHCO3, 25; D-glucose, 12.5; saturated with 95% O2/5% CO2 (pH adjusted to 7.4) at 32 °C. Slices were kept at room temperature (25 ± 1 °C) in ACSF at least for 1 h until ready for recording.
Electrophysiology
The slices were fixed in a recording chamber (∼0.4-mL volume, RC-26GLP, Warner Instruments, Hamden, CT, USA) under a nylon mesh attached to a stainless steel anchor, then submerged in, and continuously perfused with, ACSF at a flow rate of 2 mL/min. All experiments were performed at room temperature (25 ± 1 °C). The fEPSPs were recorded from the stratum radiatum (SR) in the CA1 field using borosilicate glass electrodes (3 to 5 MΩ) filled with perfusing ACSF. Recordings were made using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA) with a high-cut filter at 2 kHz.
A stainless steel concentric bipolar electrode (Unique Medical, Tokyo, Japan) was placed on the SR, and the Schaffer collateral–commissural fibers (SCs) were stimulated with a 0.1-ms pulse every 30 s. At the beginning of each experiment, a stimulus–response curve was established, and the stimulus intensity was adjusted to evoke 30% to 50% of the maximal response, which fell within the stimulus intensities of 20 to 40 µA. LTP in the CA1 region was induced by single, double, or triple tetanic stimulation (TS; 0.1-ms pulse duration, 100 Hz for 1 s) to the SCs. The fEPSPs were recorded in CA1 for 60 min after the LTP induction.
Neuronal Projection of Cholinergic Efferent Terminals from MSN by Neuron Tracing
To visualize the efferent cholinergic neurons from MSN, a total of 500 nL of recombinant adeno-associated virus including green fluorescent protein (GFP) driven by syn promoter (rAAV2/1-Syn-GFP; 1.18 × 10 13 vector genomes/mL, Addgene, MA, USA) solution was injected by a glass pipette (tip diameter 25 to 30 μm) connected to a picospritzer into MSN, then the pipette was maintained in place for an additional 10 min. Mice were sacrificed 2 to 3 wk after the injection and fixed to prepare the coronal slices for microscopy, and a total of 3 mice were used.
Synthesis of Aβ Oligomer Proto-Fibrils and Thioflavin T (ThT) Assay
AβS26C (DAEFRHDSGYEVHHQKLVFFAEDVGCNKGAIIGLMVGGVV) was purchased from Medical & Biological Laboratories (MBL) Co., Ltd. (Nagoya, Japan). Following a previous report, we generated Aβ oligomer proto-fibrils. 17 In brief, AβS26C peptides were solubilized at 0.18 mg/mL in MilliQ water, diluted 1:1 with 20-mM ammonium bicarbonate (pH 8.2) to a 20-μM (with respect to monomer) peptide solution. These were incubated at room temperature for 5 d and bubbled with oxygen for 5 to 10 min each day. AβS26C peptides form dimers by disulfide cross-linking. After lyophilization, monomeric and dimeric mutant Aβ peptides were subsequently assembled into higher ordered oligomer proto-fibril-like aggregates by incubation at 37 °C for 5 d in 20 mM phosphate buffer (pH 7.4). AβS26C assembly was investigated using a 2 mM ThT (Sigma-Aldrich, St. Louis, MO, USA) binding assay, indicating β-sheet conformation. After shaking for 15 s, each 100 μL reaction sample with 15 μL of 2 mM ThT solution was monitored in real time by ThT fluorescence (Ex 440 nM and Em 480 nM). The fluorescence of each sample was assessed by the averaging of triplet reactions. In electron microscopy (EM), the conformation of oligomer proto-fibril-like aggregates was confirmed. Negative contrast EM was performed as described in a previous report. 18 In brief, samples were applied to carbon-coated grids and observed using EM (JEM-1011J, Nihondenshi, Tokyo, Japan) after staining with 2% (w/v) uranyl acetate solution (Merck, Darmstadt, Germany).
Application of Drug and Synthetic AβS26C Oligomer for In Vitro Electrophysiology
CCh (50 nM; a cholinergic agonist) and pirenzepine (0.1 µM; a selective muscarinic M1 receptor antagonist) were purchased from Sigma-Aldrich (St. Louis, MO, USA). These drugs were dissolved in ACSF and applied by gravity feed from 60 mL reservoirs bubbled with 95% O2/5% CO2, from 15 min before LTP induction to 5 min after LTP induction. In the electrophysiological experiments to assess the suppressive effect of synthetic AβS26C oligomer, 50 nM, 75 nM, or 100 nM AβS26C (with respect to monomers) in ACSF was continuously applied from 20 min before the induction of LTP until the end of recording. To assess cholinergic effects, fresh hippocampal slices were treated with 50 nM CCh solution containing synthetic AβS26C oligomer for 20 min before LTP induction.
Data Acquisition and Statistical Analysis
All data were included in the final analyses except when the baseline response changed during the experiment. The fEPSPs were sampled online at 4 kHz (PowerLab AD Instruments, Sydney, Australia), stored on hard disk, and analyzed off-line with Scope 4 and Chart 5 (AD Instruments). The maximal slope values of the initial rising phase of fEPSPs were measured to avoid contamination of voltage-dependent components as much as possible. The magnitude of LTP was calculated as a percentage of the averaged fEPSP slope values from 50 to 60 min after each TS relative to the baseline fEPSP slope values. Derived parameters were expressed as the mean ± standard error of mean (SEM) (n = number of slices, N = number of animals) and compared using one-way analysis of variance (ANOVA) followed by multiple comparisons (Bonferroni corrected multiple t test) or unpaired t test with the level of significance set at P < 0.05.
Results
Experiment 1: Contribution of Cholinergic Mechanisms to Enhancement of LTP in CA1
Variations in LTP determined by the number of preconditioning tetanic stimuli
We previously reported that cholinergic neuronal stimulation could enhance the fEPSP slopes in hippocampal LTP, via muscarinic M1 receptor, with single TS as preconditioning. 16 In past reports, the different natures of the preconditioning stimuli, that is, tetanus or theta burst, have been shown to influence LTP enhancement through different types of glutamate receptors. 19 Our first question was whether the degree of preconditioning by TS could influence the induction of LTP. To examine the effect of glutamatergic activity on activity-dependent plasticity in pyramidal neurons, LTP in the SR of the CA1 region was investigated. After recording stable fEPSPs in the CA1 region, TS (100 Hz for 1 s) was applied to the SCs. To change the grade of glutamatergic activity inducing LTP, we used preconditioning single, double, or triple TS on SCs. At 50 to 60 min after single TS, fEPSP slopes increased to 118.6% ± 1.9% (n = 5, N = 3) of the baseline, indicating LTP induction (Fig. 1). In comparison with the fEPSP slope after single TS, those after double or triple TS were markedly enhanced to 150.6% ± 4.9% (n = 11, N = 5; P < 0.01) or 142.2% ± 1.8% (n = 7, N = 3; P < 0.05), respectively (Fig. 1). Triple TS did not enhance fEPSP slopes beyond that achieved with double TS. These results suggest that the strength of LTP depends on the degree of glutamatergic activity preconditioning and is saturated at a fixed level.
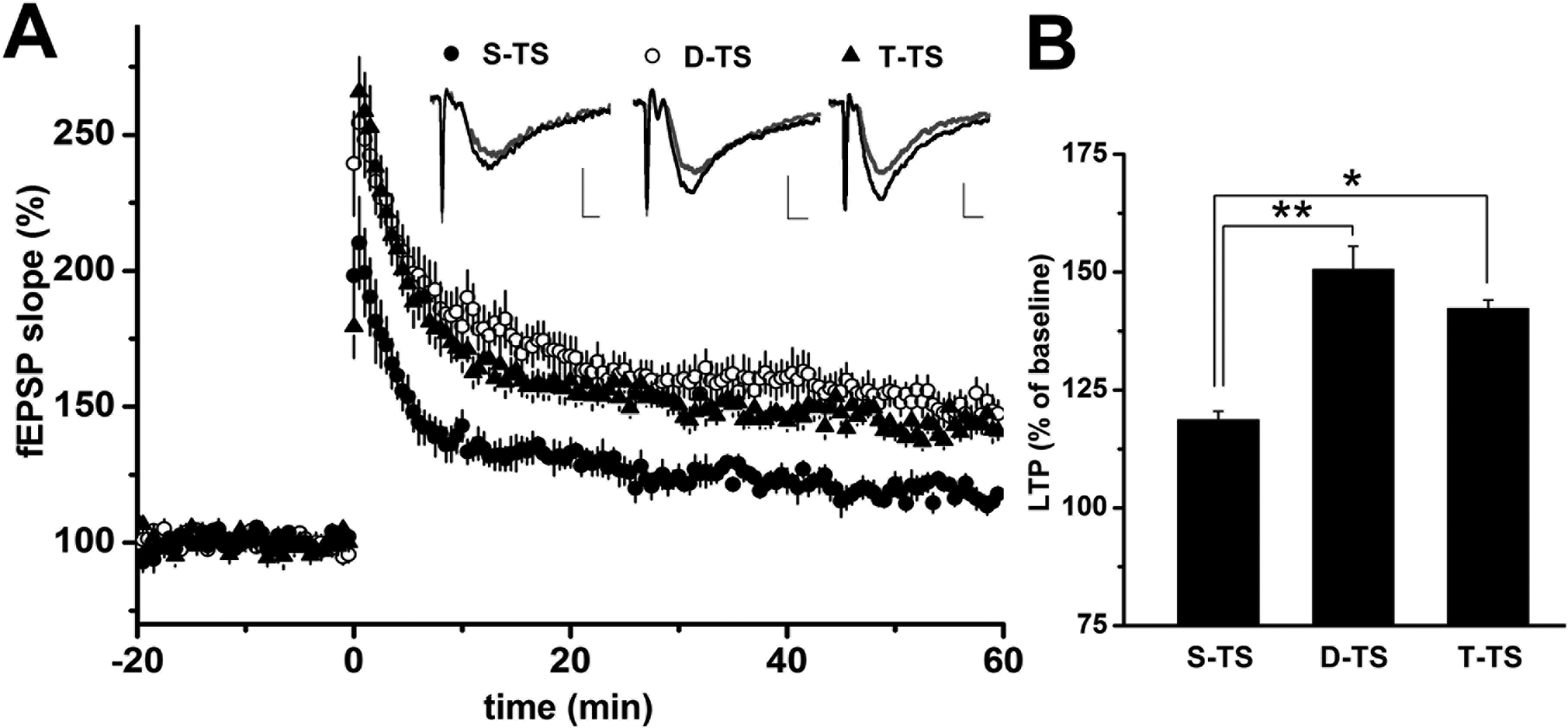
The effect of the number of preconditioning tetanic stimulations on long-term potentiation (LTP) amplitude. Time courses of field excitatory postsynaptic potentials (fEPSPs) are shown in (A). Tetanic stimulation (TS; 0.1 ms, 100 Hz for 1 s) was applied to the Shaffer collateral-commissural fibers (SCs) at time 0 with the repetitions indicated (filled circles; S-TS; single TS, n=5, open circles; D-TS; double TS n-11, filled triangles; T-TS; triple TS n=7). Representative traces of fEPSP were taken from 0 to 10 min before TS (gray line) or 50 to 60 min after TS (black line). When measured at 50 to 60 min after TS, LTP induced by D-TS or T-TS was significantly enhanced in comparison with that induced by S-TS (A,B). However, LTP induced by T-TS was not enhanced beyond that of D-TS. Data are expressed as the mean ± SEM. *P < 0.05. **P < 0.01.
The effect of cholinergic modulation on different degrees of LTP
Numerous reports have demonstrated that cholinergic stimulation may modulate glutamatergic activity-dependent plasticity via nicotinergic and/or muscarinergic receptors in LTP and its converse, long-term depression. 20 –22 Our next question was whether the cholinergic modulating system plays an important role in the enhancement of LTP resulting from single and double TS. To examine the difference in cholinergic modulation depending on the degree of glutamatergic preconditioning on LTP, we investigated the cholinergic modulating effect by continuous application of CCh (50 nM) from 15 min before LTP induction by single TS to 5 min after LTP induction. The application of CCh enhanced the fEPSP slope (136.0% ± 4.5%, n = 7, N = 5) when compared with control (118.6% ± 1.9%, n = 5, N = 3) following single TS (P < 0.05; Fig. 2). This enhancement was extinguished by pirenzepine (0.1 µM; 122.9% ± 5.2%, n = 5, N = 3), suggesting this modulating effect on LTP via muscarinic M1 receptors. In contrast, the enhancement of fEPSP slope by CCh was not observed under double TS LTP induction (140.0% ± 10.5%, n = 11, N = 5) compared to that without CCh (150.6% ± 4.9%, n = 11, N = 5; Fig. 3).
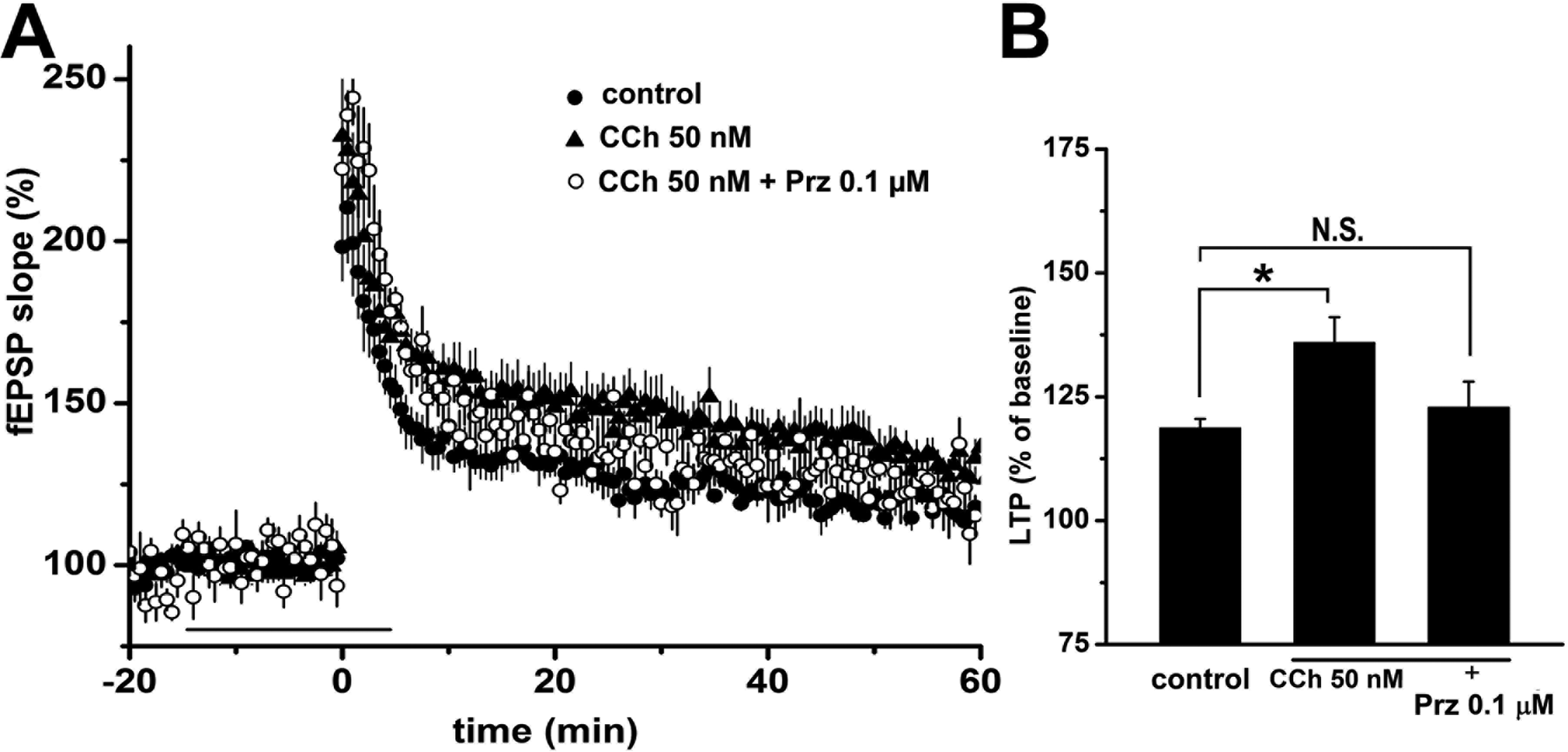
Cholinergic modulation of LTP induced by S-TS. Prior application of 50 nM carbachol (horizontal bar) significantly enhances fEPSP slope in LTP induced by S-TS (A). Simultaneous administration of pirenzepine (Prz, 0.1 µM), a M1 receptor antagonist, blocks the enhancement by carbachol, indicating muscarinic modulation of LTP enhancement in this condition (filled circles; control n = 5, filled triangles; carbachol [CCh] 50 nM n = 7, open circles; CCh 50 nM + Prz 0.1 µM n = 5). Bar graphs show the percentage change in LTP at 50 to 60 min after S-TS (B). Data are expressed as the mean ± SEM. *P < 0.05.

No modulating effect of cholinergic activity on LTP induced by D-TS. No significant enhancement of fEPSP slope was seen with prior application of CCh, (50 nM; horizontal bar), in D-TS-induced LTP. Pirenzepine (Prz, 0.1 µM) depressed LTP to a level similar to the control induced by single tetanic stimulation (S-TS; filled circles; control n = 11, open circles; CCh 50 nM n = 11, open triangles; Prz 0.1 µM n = 10; A). Bar graphs show the percentage change in LTP at 50 to 60 min after D-TS (B). Data are expressed as the mean ± SEM. *P < 0.01.
Next, to examine the involvement of cholinergic activity in the enhancement of fEPSP slope from single and double TS, we applied pirenzepine (0.1 µM) before LTP induction by double TS. The fEPSP slope was significantly reduced to a level similar to that resulting from single TS (122.1% ± 3.0%, n = 10, N = 4; P < 0.05; Fig. 3). However, the application of atropine, a nonspecific muscarinic antagonist, did not reduce the fEPSP slope induced by single TS (data not shown, see Ref. 16 ). These data suggest that cholinergic activation via muscarinic M1 receptors play a crucial role in the enhancement of fEPSP from single to double TS, and LTP induced by double TS represents the maximum postsynaptic glutamatergic activity possible. This may involve full translocation of α-amino-3-hyrdoxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors within the postsynaptic button and cholinergic neuronal activity through muscarinic M1 receptor leading to the enhancement of fEPSP slopes in double TS, but not in LTP induced by single TS.
Neuronal projection of cholinergic efferent terminals from the MSN to the SR
Our next question was whether cholinergic efferent terminals project from the MSN to the SR of CA3–CA1 and the SCs. We stimulated preglutamatergic neurons on the SCs and recorded fEPSPs in the SR of the CA1 region. Previous reports have demonstrated that the main region of cholinergic projection from MSN might be the stratum oriens in CA1, the stratum oriens in CA3, and the hilus of the dentate gyrus. 21 To verify our finding of cholinergic activity in the enhancement of LTP by double TS, we confirmed the existence of neuronal projections of cholinergic efferent terminals from the MSN in the SR of CA3–CA1 and SCs by a neuronal antegrade tracing method using AAV-GFP-injected into the MSN. Efferent synaptic terminals were observed in the SR of CA3–CA1 as well as in both the stratum oriens and the hilus of the dentate gyrus (Fig. 4).

Cholinergic projections from the medial septal nucleus (MSN) to the stratum radiatum (SR). Neuronal tracing by injection of recombinant adeno-associated virus into the MSN reveals the innervation to the SR (A). Efferent terminals from the MSN may project to postsynapses in the SR of CA1(b)–CA3(a), where the recording and stimulating electrodes, respectively, were placed as shown in schematic diagram (B).
Experiment 2: Protective Effect of Cholinergic Modulations against the Synthetic Aβ Oligomer-Induced Suppression of LTP in CA1
Suppressive effect of synthetic AβS26C oligomer on LTP
Our data suggest that cholinergic activity can modulate and enhance LTP in unsaturated postsynaptic glutamatergic neurons. We next investigated whether cholinergic activity could also modulate LTP when the postsynaptic glutamatergic activity is suppressed by an exogenous factor, such as synthetic AβS26C oligomer.
To morphologically confirm the alteration from dimeric AβS26C to proto-fibrils, we observed the incubated AβS26C using a transmission electron microscope (TEM). Negative contrast EM showed that dimeric AβS26C changed to chain-like, proto-fibril-like structures 5 d after incubation and remained stable (Fig. 5A). We also observed that these proto-fibril-like structures were ThT positive, indicating that they contain β-sheets, and the ThT-positive intensity gradually increased over 5 d (Fig. 5B). These results suggest that dimeric AβS26C can change to proto-fibril-like structures containing β-sheets, as seen in nonsynthetic Aβ oligomers. 17

Aggregated amyloid oligomers biologically suppress synaptic activity in LTP. In negative contract transmission electron microscope (TEM), mutant amyloid monomer (AβS26C) morphologically aggregates, in a time-dependent fashion, when incubated at 37 °C. Proto-fibril-like assemblies were observed after incubation for 5 d (A). Thioflavin-T binding fluorescence analysis revealed a significant increase in intensity after incubation for 5 d (P < 0.01), indicating the presence of β-sheet rich structures (n = 4 in each group; B). In LTP induced by S-TS in wild-type mice, proto-fibril-like assemblies, applied as indicated by the horizontal bar, significantly suppressed fEPSP slope in a concentration-dependent manner, indicating synaptic toxicity on the glutamatergic neural network. Representative traces of fEPSP were taken from 0 to 10 min before tetanic stimulation (TS; gray line) or 50 to 60 min after TS (black line; C). Percentage changes in LTP at 50 to 60 min after S-TS (D). Data are expressed as the mean ± SEM. *P < 0.05. NS, not significant.
We then confirmed the suppressive effect of synthetic AβS26C oligomers on LTP in the hippocampus of WT mice at 12 to 15 wk old. Synthetic AβS26C oligomers were perfused at a concentration of 50 nM, 75 nM, or 100 nM, calculated by the amount of AβS26C monomer. We evaluated the inhibitory effect of synthetic AβS26C oligomers in LTP induced by single TS. Twenty minutes after the application, the induction and evaluation of LTP were started. The fEPSP slope was suppressed by 100 nM synthetic Aβ oligomers (105.8% ± 3.9%, n = 6, N = 4; P < 0.05 vs. control) compared with control (122.7% ± 2.4%, n = 10, N = 7). Two lower concentrations of synthetic Aβ oligomers had no significant effect (50 nM: 117.8% ± 4.2%, n = 6, N = 4, P > 0.05 vs. control, 75 nM: 110.6% ± 8.6%, n = 4, N = 2, P > 0.05 vs. control; Fig. 5C and D). This suggests that synthetic Aβ oligomers can suppress the fEPSP slope enhanced by single TS in a concentration-dependent manner.
Cholinergic rescue of the fEPSP slope suppression by synthetic AβS26C oligomers
We next investigated whether cholinergic stimulation could rescue fEPSP slopes from suppression by 100 nM synthetic AβS26C oligomers. The effects of co-application of 50 nM CCh and 100 nM synthetic AβS26C oligomers to hippocampal slices 20 min before LTP induction were investigated (Fig. 6). The CCh-induced enhancement of fEPSP slope following single TS was not suppressed by synthetic AβS26C oligomers (CCh: 127.0% ± 6.0%, n = 5, N = 4, CCh with AβS26C: 125.1% ± 4.5%, n = 4, N = 3). This suggests that the exogenous cholinergic stimulation can rescue glutamatergic neuronal activity from functional suppression by synthetic AβS26C oligomers.
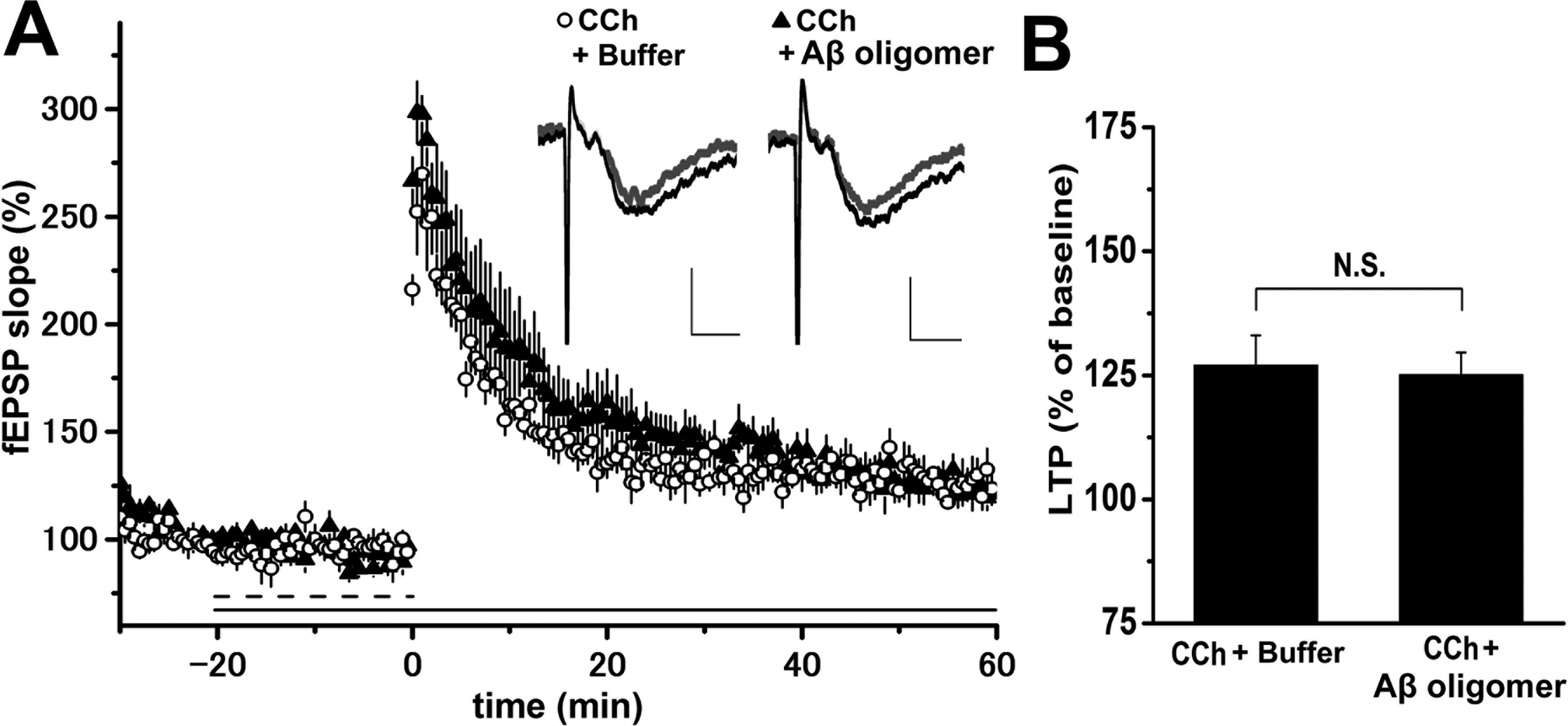
Cholinergic activation through muscarinic receptors rescues glutamatergic activity in the presence of synthetic amyloid-β (Aβ) oligomers in wild-type mice. No significant change in prior application of carbachol (dashed bar) was shown in long-term potentiation (LTP) in the presence of synthetic Aβ oligomers (horizontal bar) when compared with that without synthetic Aβ oligomers. This suggests that carbachol rescues glutamatergic activity influenced by synaptic toxicity by synthetic Aβ oligomers (filled triangles; 50 nM carbachol [CCh] + 100 nM Aβ n = 4, open circles; 50 nM CCh + buffer n = 5; A, B). Representative traces of field excitatory postsynaptic potential were taken from 0 to 10 min before tetanic stimulation (TS; gray line) or 50 to 60 min after TS (black line). Percentage changes in LTP at 50 to 60 min after TS. Data are expressed as the mean ± SEM (B). NS, not significant.
We had previously generated CaM Kinase II promoter-driven HCNP-pp transgenic (HCNP-pp Tg) mice. In these, the amount of choline acetyltransferase in the MSN is specifically increased, 23 and the fEPSP slopes of LTP induced by single TS are enhanced via M1 muscarinic modulation in the hippocampus at 12-wk-old. 16 Our final question was whether HCNP-pp could rescue fEPSP slopes in LTP induced by single tetanus following suppression by 100 nM synthetic AβS26C oligomers. To investigate this, fEPSP slopes were evaluated using hippocampal slices from HCNP-pp Tg mice. No suppression of fEPSP slope by 100 nM synthetic AβS26C oligomers was seen in HCNP-pp Tg mice (HCNP-pp Tg with Aβ oligomers: 143.0% ± 4.9%, n = 7, N = 4, HCNP-pp Tg alone: 140.1% ± 7.0%, n = 7, N = 4; Fig. 7). These data suggest that HCNP, as well as being a cholinergic stimulant, can prevent fEPSP slope suppression by synthetic AβS26C oligomers in hippocampal LTP.

Overexpression of hippocampal cholinergic neurostimulating peptide precursor protein (HCNP-pp) prevents long-term potentiation (LTP) suppression by synthetic amyloid-β (Aβ) oligomers. In LTP induced by single tetanic stimulation (S-TS) in HCNP-pp Tg mice, application of synthetic Aβ oligomers (horizontal bar) had no significant effect on the field excitatory postsynaptic potential (fEPSP) amplitude, indicating a protective effect of hippocampal cholinergic neurostimulating peptide (HCNP) against Aβ synaptic toxicity in glutamatergic neural activation (A, B). Representative traces of fEPSP were taken from 0 to 10 min before tetanic stimulation (TS; gray line) or 50 to 60 min after TS (black line). Percentage changes in LTP at 50 to 60 min after TS. Data are expressed as the mean ± SEM (B). NS, not significant.
Discussion
In this study, we demonstrated that (1) a single preconditioning TS induced unsaturated LTP, which showed a small increase in the fEPSP slope. Double and triple TS caused a further enhancement of the fEPSP slope, but the magnitude of the fEPSP slopes was similar between these stimuli, indicating saturation of LTP at this level of stimulation, (2) the cholinergic neuronal activity through muscarinic M1 receptors produced the enhancement of fEPSP slope seen between single and double TS; (3) synthetic Aβ oligomers, such as AβS26C, suppressed enhancement of fEPSP slope in unsaturated LTP induced by single TS in a concentration-dependent manner; (4) HCNP, as well as being a cholinergic stimulant, prevented the suppression of fEPSP slope by synthesized Aβ oligomers in hippocampal LTP.
The induction of LTP by high-frequency TS is used to assess the pharmacological mechanisms of synaptic function. 24 The magnitude of LTP is sensitive to several experimental variables and differs between individual experiments. Past reports have demonstrated that the type of preconditioning stimulation, that is, the frequency or intensity of stimulation, can induce different magnitudes of LTP enhancement through different types of glutamate receptors. 25,26 High-frequency TS immediately exceeds the LTP induction threshold. However, preconditioning low-frequency stimulation can suppress the magnitude of LTP subsequently induced by high-frequency stimulation, through activation of inositol 1,4,5-triphosphate receptors. 27 The magnitude of LTP typically also increases in parallel with tetanus intensity, whereas strong tetanization, that is high-intensity current, sometimes results in a paradoxical depression of fEPSP slope. 25 To indicate the enhancing effect of HCNP on LTP, we used single high-frequency TS as described in a previous report. 16 In this experimental condition, single high-frequency TS did not exceed the saturated fEPSP slope. In this study, to determine the effect of the number of preconditioning TS needed to produce saturated LTP, we investigated single, double, and triple TS on SCs as preconditioning factors. We found that LTP strength depends on the degree of preconditioning glutamatergic activity and is saturated by double high-frequency TS. However, the precise mechanism underlying this enhancement is still not understood.
We also investigated the involvement of cholinergic activation, through muscarinic M1 receptors, in the difference in enhancement between single and double TS. Cholinergic neurons originated from MSN mainly project and innervate both CA1 and CA3 SR in hippocampus,
28,29
where recording and stimulating electrodes were placed in these experiments, respectively. The modulating effect of cholinergic activity on hippocampal synaptic plasticity may be concentration-dependent. A low concentration of CCh (50 nM) can enhance fEPSP slopes induced by single TS via muscarinic M1 receptors, whereas excess acetylcholine concentration suppresses LTP.
30,31
LTP induced by TS is dependent on the activation of N-methyl-
Soluble Aβ fibril accumulation is thought to be an initial change in synaptic dysfunction and memory loss in AD. 35 Soluble Aβ oligomers impair AMPA receptor trafficking on synaptic membranes through CaMKII signaling. 36 Synthetic Aβ dimers composed of the mutant Aβ1-40, AβS26C, forms stable proto-fibril-like assemblies including β-sheets. These assemblies potently inhibit LTP in the mouse hippocampus. 17
Glutamatergic activation in the hippocampus is modulated by several neuronal networks including adrenergic and cholinergic neurons. β2-adrenergic neurons influence LTP through modulating AMPA receptors and N/L-type Ca2+ channels. 37 Enriched environments prevent the impairment of hippocampal LTP through β2-adrenergic signals, when LTP is incompletely suppressed by synthetic Aβ oligomers. 38 We previously reported that cholinergic stimulation by CCh enhances LTP through muscarinic M1 receptors, when LTP is induced by single TS. 16 Muscarinic stimulation contributes to the modulation of LTP in association with T-type Ca2+ channels. 37 In this study, we suggest that stimulation of muscarinic M1 receptors can, at least in part, prevent LTP impairment by synthetic Aβ oligomers. This result is consistent with the effects of the cholinesterase inhibitor, donepezil, for patients with AD in clinical settings.
The present study also demonstrated that HCNP overexpression could prevent LTP impairment induced by synthetic Aβ oligomers. Our previous study demonstrated that LTP in HCNP-pp Tg mice was significantly enhanced relative to that in WT mice and that this enhancement was mediated by muscarinic M1, but not nicotinic, mechanisms. Therefore, the improvement of Aβ oligomer-induced LTP impairment by HCNP overexpression may be derived from cholinergic mechanisms. This finding might clinically suggest a new kind of thinking for drug development.
As a limitation of this study, we could not directly determine the increase in acetylcholine synthesis in the hippocampus of HCNP-pp Tg mice to investigate our hypothesis that acetylcholine modulates synaptic plasticity and its suppression by synthetic Aβ oligomers through muscarinic M1 receptors. Additionally, HCNP-pp inhibits rapidly accelerated fibrosarcoma (Raf) and is therefore also referred to as Raf kinase inhibitory protein. HCNP-pp is also an ATP-binding protein and a phosphatidylethanolamine-binding protein. However, the present study could not demonstrate the contribution of these functions to prevent synthetic AβS26C oligomers from depressing LTP in HCNP-pp Tg mice, while HCNP-pp may play an important role in cerebellar long-term synaptic depression by mediating protein kinase C (PKC)-dependent mitogen-activated protein kinase (MAPK) activation. 39 In the future, further experiments are needed to elucidate the function of HCNP and/or HCNP-pp, as well as cholinergic function, in a modulating effect on synaptic plasticity in the hippocampus, including behavioral effects.
In conclusion, we demonstrate that (1) preconditioning TS can induce differential degrees of LTP depending on the number of TS delivered and that the degree of LTP induction possible is fixed to a maximum level and (2) HCNP/HCNP-pp, as well as a cholinergic stimulant, can prevent the fEPSP slope suppression induced by synthetic Aβ oligomers in the hippocampal LTP glutamatergic neuronal activity.
Footnotes
Authors’ Note
Toyohiro Sato and Yoshiaki Ohi are equally contributed to this work.
Ethical Approval
These experiments were approved by the Animal Care and Use Committee of Nagoya City University Graduate School of Medical Sciences and conformed to guidelines for the use of laboratory animal published by Japanese government (Law No. 105, October 1973).
Statement of Human and Animal Rights
All animals were kept under pathogen-free conditions on a 12 h light/dark schedule (light on 07:00 to 19:00) and given free access to food and water. Mice were deeply anesthetized with halothane before decapitation.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: N.M. was funded from Daiichi Sankyo Company (Japan), Otsuka Pharmaceutical Co., Ltd. (Japan), Sumitomo Dainippon Pharmaceutical Co., Ltd. (Japan), Merck Sharp & Dohme (MSD-Japan).