
Abstract
Hypoxia is a common cause of kidney injury and a major issue in kidney transplantation. Mitogen-activated protein kinases (MAPKs) are involved in the cellular response to hypoxia, but the precise roles of MAPKs in renal cell reactions to hypoxic stress are not well known yet. This work was conducted to investigate the regulation of extracellular signal-regulated kinase-1 and -2 (ERK1/2) and p38 and their signaling-relevant molecules in kidney epithelial cells exposed to prolonged hypoxia. Rat kidney epithelial cells Normal Rat Kidney (NRK)-52E were exposed to hypoxic conditions (1% O2) for 24 to 72 h. Cell morphology was examined by light microscopy, and cell viability was checked by 3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxypheny]-2-[4-sulfophenyl]-2H-tetrazolium (MTS). The expression of ERK1/2 and p38 MAPK, as well as their signaling-related molecules, was measured by Western blot and real-time polymerase chain (RT-PCR) reaction. At the 1% oxygen level, cell morphology had no appreciable changes compared to the control up to 72 h of exposure under light microscopy, whereas the results of MTS showed a slight but significant reduction in cell viability after 72 h of hypoxia. On the other hand, ERK1/2 and p38 phosphorylation remarkably increased in these cells after 24 to 72 h of hypoxia. In sharp contrast, the expression of transcription factor B-cell lymphoma 6 (Bcl-6) was significantly downregulated in response to hypoxic stress. Other intracellular molecules relevant to the ERK1/2 and p38 signaling pathway, such as protein kinase A, protein kinase C, Bcl-2, nuclear factor erythroid 2-related factor 2, tristetraprolin, and interleukin-10(IL-10), had no significant alterations after 24 to 72 h of hypoxic exposure. We conclude that hypoxic stress increases the phosphorylation of both ERK1/2 and p38 but decreases the level of Bcl-6 in rat kidney epithelial cells.
Introduction
The imbalance between oxygen supply and consumption leads to renal tissue hypoxia, which is the central player and final pathway to end-stage renal disease 1 –3 and occurs in acute kidney injury, chronic kidney disease, and renal cancer. 4 –6 In pathology, hypoxic stress increases pro-inflammatory cytokines and leads to inflammatory cell infiltration and inflammatory injury. 7 Hypoxia is also an acknowledged pathway to renal injury and ischemia–reperfusion, which is an inevitable event accompanying renal transplantation and is considered a common cause for delayed graft function and acute renal failure. 8 –11 Understanding the mechanisms involved in hypoxic pathophysiology is of great importance for the prevention and treatment of hypoxic renal injury, especially for improving renal allograft outcome in the recipient after kidney transplantation. 12 Hypoxia can activate a large range of signaling networks and regulate the expression of a variety of growth factors, cytokines, and signaling molecules in a cell- and tissue-specific manner. Among the large array of signaling pathways activated in the kidney, mitogen-activated protein kinase (MAPK) pathways have been frequently studied in the past.
The MAPKs are a family of serine/threonine kinases activated by growth and stress factors. Extracellular signal-regulated kinase-1 and -2 (ERK1/2), p38 MAPK, c-Jun N-terminal kinase (JNK), and ERK5/BMK1 are 4 major types of MAPKs in mammalian cells. 13,14 Activated MAPKs regulate many cellular processes such as mitosis, gene expression, metabolism, cell survival/death, and proliferation in multiple cell types. However, their expression and specific role, especially ERK1/2 and p38, in the hypoxic kidney are not fully understood yet with the existence of contradictory debates. For example, most studies suggested that p38 is a mediator in the production of pro-inflammatory cytokines 15 and in pro-apoptotic signaling, 16,17 whereas ERK1/2 is a survival modulator under hypoxic stress. 18 –20 However, reports have also suggested that kidney hypoxic injury can be combated by the inhibition of the ERK1/2 pathway. 21,22 The roles of p38 under hypoxic conditions are also controversial, similar to ERK1/2. Most studies have suggested that the activation of p38 is an apoptotic signal to cells under hypoxic/ischemic conditions. 16,23,24 On the other hand, it was indicated that the activation of p38 pathways is able to ameliorate renal hypoxic injury and inflammatory responses. 25,26 It is important to clarify this issue because renal hypoxia has pivotal roles in the development and progression of acute and chronic kidney disease, renal cancer, as well as kidney transplantation. It is possible to find useful clues for new therapeutic treatments for hypoxic kidney diseases by targeting MAPKs after better understanding their function and mechanisms in hypoxic conditions.
Our previous studies in the cortical neurons showed that ERK1/2 and p38 were differentially involved in neuronal responses to hypoxic stress. The cross-talk between ERK1/2 and p38 displays a “yin-yang” antagonism under the control of protein kinase C (PKC) pathway. 27 Therefore, we applied the same hypoxic stress to rat kidney epithelial (NRK-52E) cells for 24 to 72 h and then detected the changes in the expression and phosphorylation of ERK1/2 versus p38 and PKC, as well as other relevant signaling molecules, to define the hypoxic changes in ERK 1/2 versus p38 and compare the difference between neuronal and kidney epithelial cells.
Materials and Methods
Cell Culture
NRK-52E cells were purchased from The Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. Briefly, NRK-52E cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) containing 5% fetal bovine serum (FBS) (Hyclone, Logan, UT, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin (Gibco) and incubated in a humidified atmosphere with 5% CO2 at 37 °C.
Establishment of Hypoxic Culture Condition
A hypoxic incubator was used to expose cells to continuously controlled hypoxic conditions. This humidified and temperature-controlled (37 °C) chamber was supplemented with 5% CO2 and N2 (as required to maintain controlled O2 levels). Cells were exposed to 1% O2 for varying durations. In parallel, normoxic controls were incubated in a humidified incubator supplemented with 21% O2 and 5% CO2 at 37°C.
Light Microscopy
Cell morphology was determined by light microscopy to ascertain the effect of hypoxic injury on cell outcome. Cells were seeded in 6-well plates, and the monolayers were examined for morphological changes that would be indicative of survival or cell death. Survival was determined by the retention of an intact monolayer and no change in cell morphology. Cell death was assigned to monolayers that detached in sheets or when a significant amount of individual cells were detached from the plate.
Cellular Proliferation Assay
The NRK-52E cells (1 × 104 cells/well) were seeded in a 96-well plate and incubated in normal culture condition for 24 h before hypoxia. Cell proliferation was measured using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxypheny]-2-[4-sulfophenyl]-2H-tetrazolium [MTS]) kit (Promega, Madison, WI, USA), according to the manufacturer’s instructions. MTS reagent (20 µL) was added to the cells in each well followed by incubation for 1 h in a 37 °C, 5% CO2 humidified incubator. The absorbance was measured at the wavelength of 490 nm using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). In accordance with the manufacturer’s instructions, the first strand of complementary DNA (cDNA) was synthesized using the PrimeScript First Strand cDNA Synthesis Kit (Takara Bio, Inc., Dalian, China) in a reaction mixture with a final volume of 20 μL, containing 1 μg total RNA, 4 μL 5× PrimeScript buffer, 1 μL deoxynucleotide triphosphate mixture, 1 μL Oligo (dT) primer, 0.5 μL PrimeScript RTase, 0.5 μL RNase inhibitor, and RNase-free water. (All these reagents/kits were from Takara Bio.) The reverse transcription reaction was performed under the following conditions: 42 °C for 15 min, followed by a termination step at 95 °C for 2 min. The standard reaction volume was 25 μL, containing 1 μL SYBR Green PCR Master Mix (Takara Bio, Inc.), 2 μL cDNA template, and 0.25 μM forward and reverse primers. The initial PCR step was as follows: 2 min at 50 °C, followed by a 15-min hold at 95 °C. This was followed by 40 cycles, consisting of a 15-s denaturation step at 95 °C, a 20-s annealing/extension step at 59 °C, and a 72 °C incubation step for 20 s. All reactions were performed in triplicate. Following normalization to the β-actin gene, the expression levels for each target gene were analyzed using the comparative threshold (CT) cycle method. The 2ΔΔCT was calculated to determine the relativity using the following formula:
Primers for Real-Time PCR.

Abbreviations: ERK1, extracellular regulated protein kinase-1; ERK2, extracellular regulated protein kinase-2; PCR, polymerase chain reaction.
Protein Extraction and Western Blot Analysis
Cells were washed twice with ice-cold phosphate-buffered saline (PBS), solubilized in lysis buffer (KeyGen Biotech Co., Ltd., Nanjing, China) on ice, and then quantified using the bicinchoninic acid assay (BCA) method (KeyGen Biotech Co., Ltd.). The preparation of nuclear extracts from the rat kidney cells was performed using the nuclear extract kit (also from KeyGen Biotech Co., Ltd.). Cell lysate proteins (50 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Solarbio Science & Technology Co., Ltd., Beijing, China) and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% nonfat milk (Bio-Rad Laboratories) in Tris-buffered saline Tween-20 (TBST; Solarbio Science & Technology Co., Ltd) buffer for 1 h at room temperature and incubated overnight at 4 °C with the indicated primary antibodies. β-actin (1:3,000), phospho-ERK1/2 (1:1,000), ERK1/2 (1:1,000), phospho-p38 (1:1,000), p38 (1:1,000), protein kinase A (PKA; 1:1,000), PKC (1:1,000), and B-cell lymphoma 2 (Bcl-2; 1:500) were purchased from Cell Signaling Technologies (Danvers, MA, USA); rabbit polyclonal anti-tristetraprolin (TTP; 1:200) and Bcl-6 (1:100) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); nuclear factor erythroid 2-related factor 2 (Nrf-2; 1:1,000) and interleukin-10 (IL-10; 1:1,000) were purchased from Abcam (Cambridge, UK). After the membranes were washed with TBST buffer, they were reacted with the appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies for 1 h at room temperature. After extensive washing with TBST buffer, the proteins were visualized with enhanced chemiluminescence reagent (Thermo Fisher Scientific, Rockford, IL, USA). All the signals of Western blots were captured by film, and band densities were quantified using Quantity One (Bio-Rad Laboratories) software and values were corrected to actin levels.
Statistics
Statistical analyses were performed using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA). Results were expressed as the mean (SEM) ± standard error of the mean of data obtained from at least 3 independent experiments. Analysis of variance (ANOVA) was utilized to analyze the difference between various groups. P < 0.05 was considered statistically significant.
Results
Effect of Hypoxia on Cell Viability/Injury
We have previously shown that hypoxia (1% O2) for 48 to 72 h caused severe neuronal injury with an upregulation of p38 signaling. 27 We therefore investigated the viability/injury of hypoxia-exposed kidney epithelial cells by morphological examination and MTS assay. Under light microscopy, the morphology of hypoxic cells had no appreciable changes as compared to those in normoxic conditions (Fig. 1A). The MTS assay did not detect any significant difference in cell viability between the hypoxic and normoxic cells in the first 48 h (P > 0.05). There was only a slight decrease in the cell viability after 72 h of hypoxic exposure (from 164.07% ± 7.93% in normoxia to 143.10% ± 3.93% in hypoxia, P < 0.05, n = 3; Fig. 1B). Since the hypoxic condition was the same as in our previous study on neuronal cells, 27 the data suggest that kidney epithelial cells are more tolerant to hypoxic insults than neuronal cells.

Morphology and viability of the rat kidney epithelial cells (NRK-52E) exposed to hypoxia. After the cells were exposed to hypoxia at 1% O2 for 24, 48, or 72 h, cell morphology was examined by light microscopy (A) and the cell viability was measured by 3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxypheny]-2-[4-sulfophenyl]-2H-tetrazolium (MTS) assay. (B) At least 3 independent experiments were carried out in all groups. H, hypoxia; C, normoxic control. The photomicrographs were taken with 40× magnification (scale bar, 100 μm) and 100× magnification (scale bar, 40 μm). *P < 0.05. Note that cell morphology under light microscopy showed no appreciable difference between the control and hypoxia, while the MTS assay indicated a slight decrease in cell viability after 72 h of hypoxia.
Effect of Hypoxia on ERK1/2 and p38 Phosphorylation
Since ERK1/2 and p38 are differentially regulated in neuronal cells under hypoxia as shown in our previous work, 27 we first investigated if they behaved in a similar way in kidney epithelial cells under hypoxic conditions. Total and phosphorylated ERK1/2 and p38 proteins were measured in NRK-52E cells exposed to 24 to 72 h of hypoxia. As shown in Figure 2 (A and B), hypoxia induced a large increase in phosphorylated ERK1/2 (P-ERK1/2) in the NRK-52E cells at all 3 time points (24, 48, and 72 h) of hypoxic exposure (P < 0.001, n = 4) without any significant change in total ERK 1/2 (T-ERK1/2) or ERK1/2 messenger RNA (Fig. 3). Indeed, the ratios of phosphorylated to total ERK1/2 at 24, 48, and 72 h of hypoxia increased by 6.5-, 7.2-, and 6.6-fold, respectively, as compared to those of the normoxic cells (P < 0.001, n = 4). In parallel with the increase in phosphorylated ERK1/2, phosphorylated p38 (P-p38) also increased largely without any appreciable changes in total p38 (T-p38; Fig. 2A and C). The ratios of P-p38 to T-p38 at 24, 48, and 72 h of hypoxia increased by 2.6-, 2.3-, and 2.4-fold, respectively, as compared to those of the normoxic cells (P < 0.01 at 24 and 48 h, P < 0.05 at 72 h, n = 3). These data suggest that ERK1/2 and p38 in the kidney epithelial cells are very sensitive to O2 deprivation, with a major increase in their phosphorylation in response to hypoxic stress.

Extracellular regulated protein kinase-1 and -2 (ERK1/2) and p38 protein expression under hypoxic condition in NRK-52E cells. Rat kidney epithelial cells (NRK-52E) were exposed to hypoxia at 1% O2 for 24 to 72 h. (A) Representative Western blots of ERK1/2 and p38 protein. (B) Relative quantitation of phosphorylated ERK1/2 (P-ERK1/2)/total ERK1/2 (T-ERK1/2). (C) Relative quantitation of phosphorylated p38 (P-p38)/total p38 (T-p38). β-actin was detected as an internal standard. At least 3 independent experiments were carried out in all groups. All the blots were normalized to β-actin, which was presented as 100% control. H, hypoxia; C, normoxic control. *P < 0.05. **P < 0.01. ***P < 0.001. Note that hypoxia at 1% O2 for 24 to 72 h significantly increased the expression of phosphorylated ERK1/2 and p38 but did not induce any appreciable changes in total ERK1/2 and p38 protein in NRK-52E cells.

Extracellular regulated protein kinase-1 and -2 (ERK1/2) messenger RNA (mRNA) levels in hypoxic NRK-52E cells. The cells were exposed to hypoxia at 1% O2 for 24 h. Total mRNA was extracted from the cells and analyzed by quantitative real-time polymerase chain reaction (n = 4). H, hypoxia; C, normoxic control. Note that hypoxia did not significantly change the mRNA levels of ERK1/2.
Expression of PKC, PKA, and Bcl-2 Under Hypoxia
Since PKC and Bcl-2 are critically involved in MAPK signaling and in protection against hypoxic injury in neuronal cells, 27 we considered whether the tolerance of kidney epithelial cells toward hypoxia is related to their regulation. We therefore comparatively measured PKC, PKA, and Bcl-2 proteins under hypoxia for different durations. However, we did not see any significant changes in all these signaling proteins after hypoxia for 24 to 72 h (P > 0.05; Fig. 4). These observations suggest that kidney epithelial cells have a different response to hypoxia than neuronal cells in terms of PKC, PKA, and Bcl-2 signaling.

The protein expression of B-cell lymphoma 2 (Bcl-2), protein kinase C (PKC), and protein kinase A (PKA) under hypoxia of NRK-52E cells. The cells were exposed to hypoxia at 1% O2 for 24 to 72 h. (A) Representative Western blots of Bcl-2, PKC, and PKA proteins in whole cells. (B) Relative quantitation of Bcl-2, PKC, and PKA proteins. At least 3 independent experiments were performed in all groups. All the blots were normalized to β-actin, which was presented as 100% control. H, hypoxia; C, normoxic control. Note that there are no significant changes in Bcl-2, PKC, and PKA expression in whole cell proteins.
Nuclear level of Nrf-2 Under Hypoxia
Furthermore, we explored whether Nrf-2 was involved in the response of the kidney epithelial cells to hypoxia by detecting its protein level in nuclear proteins because its translocation and function were reported as a cytoprotective strategy against hypoxic injury in astrocytes and other cells. 28 Similarly, the Nrf-2 protein had no significant change after hypoxic exposure for 24 to 72 h (Fig. 5), suggesting that Nrf-2 is unlikely to be involved in the tolerance of the kidney epithelial cells to hypoxic stress.
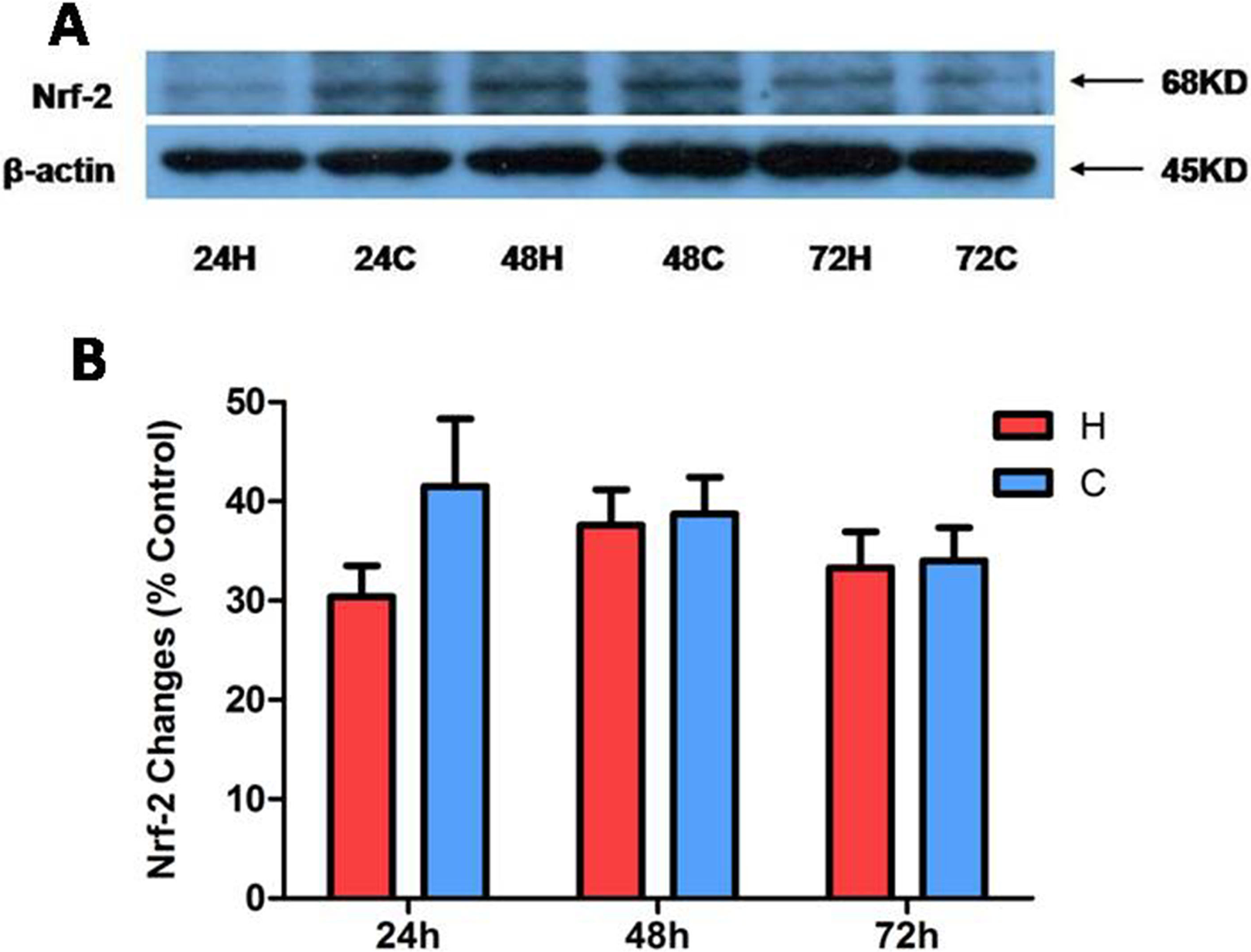
Nuclear factor erythroid 2-related factor 2 (Nrf-2) protein expression in hypoxic NRK-52E. The cells were exposed to hypoxia at 1% O2 for 24 to 72 h. (A) Representative Western blots of Nrf-2 in the nuclear fraction of rat kidney epithelial (NRK-52E) cells exposed to hypoxia. (B) Relative quantitation of Nrf-2. At least 3 independent experiments were conducted in each group. β-actin was performed as 100% loading control. H, hypoxia; C, normoxic control. Note that there are no significant changes in the expression of nuclear Nrf-2 protein.
The Expression of Bcl-6, TTP, and IL-10 After Prolonged Hypoxia in Kidney Cells
Finally, we asked if the hypoxic tolerance of kidney epithelial cells is attributed to an upregulation of anti-inflammatory factors because hypoxia triggers inflammatory reactions and thus leads to kidney injury. 29,30 We measured TTP and IL-10 proteins and potential anti-inflammatory cytokines 31 –35 using Western blots. We also detected the change in Bcl-6 protein, a transcriptional repressor, 36 in the same cell samples since Bcl-6 is relevant to MAPK signaling process. 37 Our data showed no significant changes in TTP and IL-10 after hypoxic stress for 24 to 72 h (P > 0.05, n = 3). Interestingly, there is a major decrease in Bcl-6 protein, especially after 24 and 48 h of hypoxia. Indeed, Bcl-6 protein levels decreased to 43.71% ± 5.45% and 50.18% ± 24.50% at 24 and 48 h, respectively, after hypoxic exposure (P < 0.05 vs. the control, n = 3; Fig. 6A). The data showed that Bcl-6 expression was decreased by prolonged hypoxia in NRK-52E cells. All these data suggest that Bcl-6 is involved in the process of kidney epithelial cell response to hypoxic stress.

Effects of hypoxia on tristetraprolin (TTP), interleukin-10 (IL-10), and B-cell lymphoma 6 (Bcl-6) expression in NRK-52E cells. The cells were exposed to hypoxia at 1% O2 for 24 to 72 h. (A) Representative Western blots of Bcl-6, TTP, and IL-10 proteins in whole cells; (B) Relative quantitation of Bcl-6, TTP, and IL-10 protein (n = 3). β-actin was detected as an internal standard. H, hypoxia; C, normoxic control. *P < 0.05. Note that hypoxia significantly decreased the level of Bcl-6 protein but had no impact on TTP and IL-10 in the same hypoxic condition.
Discussion
The present study has made the following findings in rat kidney epithelial cells: (1) hypoxia at 1% of O2 for 24 to 72 h only slightly injured the rat kidney epithelial cells, unlike neuronal cells which showed severe injury after prolonged hypoxia; (2) prolonged hypoxic insult not only increased p38 phosphorylation but also dramatically promoted ERK1/2 phosphorylation; and (3) under the same hypoxic conditions, Bcl-6 was significantly downregulated, while PKA, PKC, Bcl-2, Nrf-2, TTP, and IL-10 that are relevant to ERK1/2 and p38 signaling had no appreciable changes. Our novel findings suggest that ERK1/2, p38, and Bcl-6 are differentially involved in response of rat kidney epithelial cells to prolonged hypoxia, while PKA, PKC, Bcl-2, Nrf-2, TTP, and IL-10 have no significant changes in terms of their expression in response to the same hypoxic stress.
In kidney transplantation, hypoxia is a critical factor affecting the development of renal damage during the transplant process from donor to recipient. 8,12 Hypoxia is also believed to play a key role in both acute and chronic kidney diseases and in the transition from acute to chronic kidney diseases. 38 In fact, it is thought that hypoxia plays a major role in renal pathology by promoting the initial tubular damage and inducing a sustained inflammatory attack. 39 –41 Our previous studies on rat cortical neurons showed that hypoxia at 1% of O2 for 48 to 72 h caused severe neuronal injury/death. 27 In sharp contrast, the data from this work show that hypoxia at the same oxygen level for 24 to 72 h caused only very mild injuries to these kidney cells. It is our belief that rat kidney epithelial cells are very likely more tolerant to hypoxic stress than central neurons, since we used compatible hypoxic conditions for both neuronal and kidney cells. Further study is needed to verify whether other types of renal cells share the same cellular property.
MAPKs are differentially involved in the regulation of pathophysiological mechanisms for a great number of human diseases, ranging from Parkinson disease 42 to tumorigenesis. 43 Indeed, MAPKs have been implicated as an important regulator for cell survival/injury under hypoxic conditions. 44,45 Among them, the role of ERK1/2 has been considered a protective factor for cell survival, while p38 is considered a factor for cell injury. 20,46 –49 In cultured rat cortical neurons, hypoxia differentially regulated the phosphorylation of p38 and ERK1/2, 27 whereas in the rat hippocampal slices, the phosphorylation of both p38 and ERK2, but not ERK1, was upregulated in response to hypoxia. 50 It is also reported that chronic inflammatory and apoptotic cascades that are stimulated by p38 and JNK, but not ERK pathways, contribute to the pathogenesis of the chronic intermittent hypoxia-induced renal injury. 51 These studies suggest that MAPKs are differentially involved in cellular responses to hypoxic stress, depending on cell type and cell conditions. However, the precise functions of ERK1/2 and p38 are still debatable, especially in the kidney under hypoxic conditions. For example, it is reported that ERK1/2 is involved in injury and apoptosis rather than contributing to cell survival in kidney cells, 21,22 while the activation of p38 can inhibit hypoxic injury in kidney cells. 26 Our studies on cortical neurons showed that there is an increase in p38 phosphorylation and a decrease in ERK1/2 phosphorylation after prolonged exposure to severe hypoxia. 27 Interestingly, the data from the present study showed a completely different pattern that there was no decrease in ERK1/2 phosphorylation at all. In fact, ERK1/2 phosphorylation was remarkably promoted by prolonged hypoxic stress. As shown in our previous work and those of others, 18,47,52,53 the balance between the ERK1/2 and p38 signals is critical for cell survival under hypoxic stresses. Based on our observations on neurons, ERK1/2 functions as a survival signal under hypoxic conditions and there is a “yin-yang” pattern between ERK1/2 and p38 in neuronal injury and protection under hypoxia. 27 If ERK1/2 signaling is stronger than p38 activity, cells may better adapt to hypoxic environments; otherwise, cells may be prone to apoptosis. The regulatory mechanisms for ERK1/2 could be very different between kidney and neuronal cells in hypoxia because severe hypoxia inhibits its activity in neurons 27 but promotes it in kidney epithelial cells. As a result, increased ERK1/2 signaling counteracts the apoptotic effect of p38 signaling, protecting the kidney cells from hypoxic injury. This is most likely the reason why rat kidney epithelial cells are more tolerant to hypoxic insults. Indeed, we strongly believe that the upregulation of ERK1/2 signaling is a key factor for renal cells to survive in hypoxic conditions. Therefore, the dramatic upregulation (instead of downregulation as seen in neurons 27 ) of ERK1/2 is very likely an important mechanism for the tolerance of the kidney epithelial cells to hypoxic stress in this work. However, more studies are needed to clarify whether this is a general phenomenon in the kidney or a unique characteristic of this particular type of kidney cell.
Our data show that the increase in ERK1/2 and p38 phosphorylation is associated with the downregulation of Bcl-6 in hypoxic kidney epithelial cells. Bcl-6 is known to be expressed and is an active as transcription factor in germinal center B cells, mature cardiac myocytes, testis, pulmonary epithelium, and hepatocytes. 54 –58 It has been hypothesized that downregulation of Bcl-6 is required for germinal center B cells to differentiate into plasma cells. 59 Its signaling is able to enhance the expression and thereby the activity of hepatocyte nuclear factor (HNF)-1α, a transcription factor important for the regulation of numerous genes involved in several metabolic processes in the kidneys. 60 An increase in its expression can inhibit the apoptosis of lymphoma cells treated with chemotherapeutic reagents, most likely through enhancement of the antioxidant defense systems. 55 It is also indicated that inhibition of Bcl-6 function by a dominant-negative form of Bcl-6 can inhibit the cell cycle progression and induce apoptosis in Raji cells. 61
Previous studies showed that the activation of peroxisome proliferator–activated receptor (PPAR)-δ increased both total and free Bcl-6 levels and inhibited the activation of p38 and ERK1/2, 62 and the activated form of p38 was detected in spermatocytes of adult Bcl-6-deficient mice. 63 Therefore, Bcl-6 may contribute to negative regulation of MAPKs. On the other side, MAPKs may regulate Bcl-6. It was reported early in 1997 that the functions of Bcl-6 were regulated by its phosphorylation mediated by the MAPK signaling pathway in germinal center B cells. 64 Niu et al. observed that antigen receptor activation leads to (1) Bcl-6 phosphorylation though ERK activation and (2), in turn, targeted Bcl-6 for rapid degradation by the ubiquitin/proteasome pathway. 65 ERK1/2 may play an important role in the regulation of Bcl-6 abundance in the pathogenesis of Bcl through Bcl-6 phosphorylation, which can lead to Bcl-6 ubiquitination and proteasomal degradation. On the other hand, Batlle et al. showed that p38 induces Bcl-6 transcription and may increase Bcl-6 protein expression when the strong pathways repressing Bcl-6 were not activated. 37 Therefore, ERK1/2 may be the dominant regulatory factor over p38 in determining the fate of Bcl-6. Taken together, MAPKs, especially ERK1/2, may have a reciprocal inhibition with Bcl-6. The downregulation of Bcl-6 may result from the upregulation of MAPKs, particularly ERK1/2, in hypoxic kidney epithelial cells, although we cannot rule out the possibility that prolonged hypoxia directly inhibits Bcl-6.
Our work and those of others have suggested that PKC, PKA, and Bcl-2 are directly or indirectly involved in MAPK signaling in hypoxic injury or protection against hypoxic injury. 27,66 –69 For example, Bcl-2 confers protection against nuclear mitochondrial impairment under oxidative stress in pheochromocytoma (PC-12) cells, and a reduction in Bcl-2 expression leads to cell injury. 70 However, we did not find any appreciable change in the kidney epithelial cells, even after 72 h of hypoxia, which was different from what we saw in hypoxic neurons. 27 The mechanisms underlying the difference may be complex. A simple explanation was that since both ERK1/2 and p38 were upregulated, these 2 MAPKs may reach a new balance at a new level, thus keeping MAPK-relevant molecules unchanged as a result.
Inflammation and apoptosis contribute to the pathogenesis of many diseases, ranging from renal diseases 71,72 to neurological disorders. 73 –77 Hypoxia can initiate a cascade of deleterious cellular responses leading to inflammation and cell death in many circumstances. 78 –80 Since we saw that severe hypoxia had only a slight impact on kidney epithelial cells, even after a prolonged exposure, we checked the expression of 3 anti-inflammatory molecules such as Nrf-2, TTP, and IL-10 in these cells. The transcription factor Nrf-2 is a well-known regulator of cellular responses to hypoxic, ischemic, and oxidant stress for cytoprotection. 28,81,82 The anti-inflammatory molecule TTP can limit the expression of a number of critical genes for chronic inflammatory diseases and cancers. 31 –33 IL-10 was reported to play an important role in the maintenance of intestinal homeostasis by preventing inflammation 35 and by working as a cytoprotective agent involved in anti-inflammatory pathways. 34 Unexpectedly, we did not observe any significant alteration in these cytoprotective molecules. The reason may be more or less the same as the reason for protein kinases and Bcl-2, stated above. Since ERK and p38 are well balanced, cells may not necessarily mobilize more energy-consuming processes inside the cells.
On the other hand, the data on these unchanged molecules strongly support that the upregulation of ERK and p38 and downregulation of Bcl-6 are specific to kidney epithelial cells in response to hypoxic stress.
In summary, our results suggest that kidney epithelial cells are more tolerant to prolonged hypoxia with an increase in both ERK1/2 and p38 phosphorylation, along with a decrease in Bcl-6. In sharp contrast, there were no significant changes in PKA, PKC, Bcl-2, Nrf-2, TTP, and IL-10 in the same cells. The increased ERK1/2 activity may combat p38-mediated cell injury, thus rendering renal cells more tolerant to hypoxic insult. The downregulation of Bcl-6 may, at least partially, contribute to hypoxic upregulation of ERK1/2. Our data provide a new perspective for cell adaptation to a hypoxic environment and potential mechanisms behind the differences in cell tolerance to hypoxia among various cell types (e.g., renal vs. neuronal cells). We may shed a light on new targets for the treatment of hypoxic injury through more in-depth research.
Footnotes
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article, and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (#81273267 and #81574053), 973 Program (2012CB518502), Shanghai Key Laboratory of Acupuncture Mechanism and Acupoint Function (14DZ2260500), and Science and Technology Commission of Shanghai Municipality (15441903800).