
Abstract
With the advent of personalized medicine, clinical trials studying treatment effects in subpopulations are receiving increasing attention. The objectives of such studies are, besides demonstrating a treatment effect in the overall population, to identify subpopulations, based on biomarkers, where the treatment has a beneficial effect. Continuous biomarkers are often dichotomized using a threshold to define two subpopulations with low and high biomarker levels. If there is insufficient information on the dependence structure of the outcome on the biomarker, several thresholds may be investigated. The nested structure of such subpopulations is similar to the structure in group sequential trials. Therefore, it has been proposed to use the corresponding critical boundaries to test such nested subpopulations. We show that for biomarkers with a prognostic effect that is not adjusted for in the statistical model, the variability of the outcome may vary across subpopulations which may lead to an inflation of the family-wise type 1 error rate. Using simulations we quantify the potential inflation of testing procedures based on group sequential designs. Furthermore, alternative hypotheses tests that control the family-wise type 1 error rate under minimal assumptions are proposed. The methodological approaches are illustrated by a trial in depression.
1 Introduction
With the advent of personalized medicines, clinical trials studying treatment effects in subpopulations have gained more and more attention. The objective of such studies is to identify subpopulations based on biomarkers, where the treatment has a positive effect. Here the term biomarker is used in a very general sense as a synonym for a baseline patient characteristic, like demographic, clinical or genetic variables or a combination of these. They are measured prior to treatment and therefore cannot be affected by the outcome. For example, there is an extensive discussion in the literature whether biomarkers can be used to predict the treatment effect of medicines in patients with depression.1,2 Although a number of treatment options for such patients are available, no single treatment is universally effective. Biomarkers can be prognostic or predictive, where prognostic biomarkers predict the outcome in a natural cohort, and predictive biomarkers, in contrast, predict the treatment effect of an experimental treatment in comparison to a control group. 3 Note that biomarkers may be both prognostic and predictive.
A wide range of methods for the identification and confirmation of targeted subpopulations in clinical trials has been proposed. 4 Several authors focused on settings, where subpopulations are defined by a continuous biomarker which is dichotomized to define biomarker-low and biomarker-high subpopulations. The subpopulation with an expected beneficial treatment effect is called the biomarker positive subpopulation and the complementary subpopulation is called the biomarker negative subpopulation. Then, hypotheses tests to test for treatment effects in the subpopulation of biomarker positive patients and the full population are performed. Because several hypotheses are investigated, an appropriate multiple testing procedure has to be applied to control the family-wise type 1 error rate (FWER).5–7
An important problem is the choice of the threshold. To obtain a conservative hypothesis testing procedure to test for treatment effects in subpopulations, the considered threshold needs to be defined a priori, either based on an independent data set or theoretical considerations. If there is uncertainty regarding the choice of the threshold, more than one threshold may be investigated. The nested structure of subpopulations defined by different thresholds for a continuous biomarker is similar to the structure of analysis populations in group sequential trials. Hence, it has been proposed to use critical boundaries of group sequential designs 8 to test nested subpopulations.6,9 However, the validity of these designs depends on the assumption that the variance of the outcomes does not vary across subgroups.
In this paper, we show that great care has to be taken when applying group sequential boundaries to test hypotheses for multiple nested subpopulations as proposed in the literature. 9 We show that for biomarkers with a prognostic effect that is not adjusted for in the statistical model, the variability of the outcome may vary across subpopulations. As this may have an impact on the correlation of the test statistics, the use of group sequential boundaries may not guarantee control of the FWER. Using simulations, we quantify the potential inflation of the FWER of testing procedures based on such group sequential designs. To obtain test procedures that control the FWER, we show how inverse normal combination tests 10 and sequential regression tests 8 can be applied to this testing problem. Furthermore, we consider a test accounting for the different variances across subgroups 6 and propose a modification of this test that accounts for the respective degrees of freedoms of the test statistics using the quantile substitution method. We show that the latter procedure controls the FWER under minimal assumptions and compare its power under a range of scenarios to alternative approaches. In addition, we generalize the multiple t-test to general subgroup tests for non-nested subgroups. To illustrate the procedures, we give a clinical trial example in depression.
2 Statistical model and testing problem
Consider a randomized parallel group clinical trial designed to evaluate a novel treatment compared to a control with a per group sample size of n. For simplicity, equally sized groups are assumed. For each subject
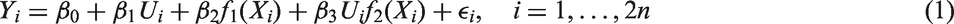
Consider an analysis strategy with the goal to identify a (sub)population, defined by a dichotomization of the biomarker Xi, where the treatment has a positive effect. To this end, we consider nested subpopulations
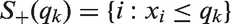
Thus, here the biomarker positive subpopulations (for which a positive treatment effect is expected) consist of all patients with biomarker values below the threshold qk (later we will also discuss the case of more general types of biomarker positive subgroups).
Separate hypotheses tests in the biomarker positive subpopulations could be considered, e.g., if there exists prior information that the treatment effect in a biomarker positive population may be larger than in the corresponding biomarker negative population; however, insufficient information on the dependence structure of the outcome on the biomarker is available and therefore several thresholds are investigated. To confirm a positive treatment effect in the considered subpopulations, we compare the mean responses of the treatment and control group of each subpopulation
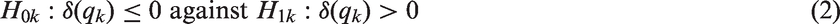
2.1 A step function model
A statistical model corresponding to the above analysis strategy can be written as a special case of equation (1). For a given threshold Dependence of the outcome on the biomarker value. (a)A step function dependence and (b) a linear dependence investigated in the simulation studies. Here, 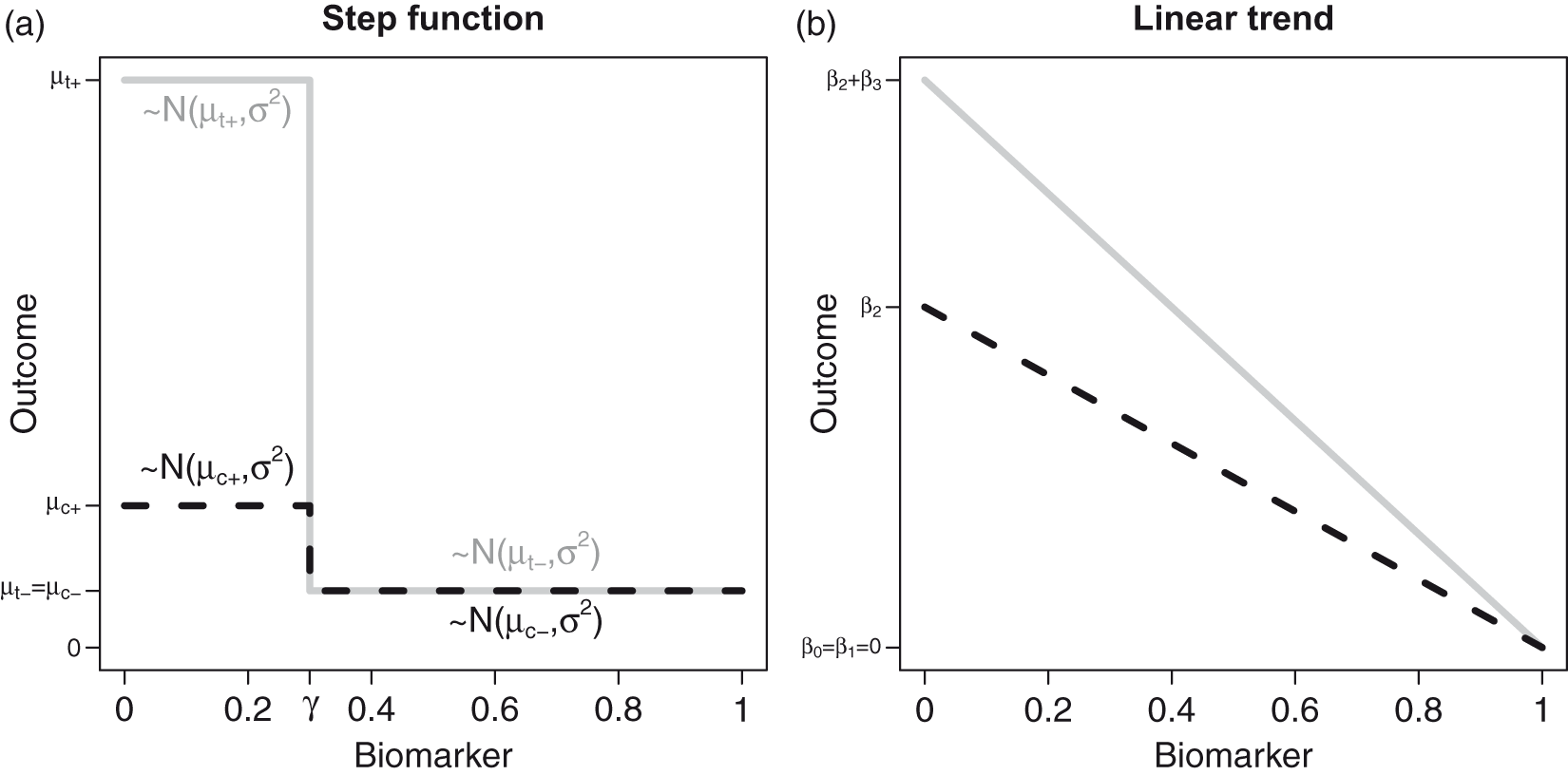

2.2 A linear trend model
An alternative model is a linear trend model, where
Note that under the global null hypothesis stating that all
3 Multiple hypotheses tests
Because multiple hypotheses are tested, the testing procedure needs to adjust for multiplicity to ensure strong control of the family-wise type 1 error rate (FWER) at pre-specified level α. A procedure controls the FWER in the strong sense if the probability that at least one true null hypothesis is rejected, is bounded by α, regardless of how many or which null hypotheses are holding. For the procedures given below, we investigate the FWER control under the global null hypothesis of no treatment effect in any of the subpopulations which implies weak FWER control only. However, strong FWER control follows by the closed testing principle since for all considered procedures it is easy to see that the rejection region for the test of hypotheses
3.1 Procedures based on multiple z- or t-tests
Assume that each hypothesis
3.1.1 The Šidák test
The Šidák test applies significance levels
3.1.2 Group sequential critical boundaries
In group sequential designs, a null hypothesis is tested repeatedly on accumulating data. Adjusted critical boundaries are applied to account for the multiple testing of the hypotheses.8,13 These boundaries are calculated while accounting for the correlation of the test statistics. Because the nested structure of the analysis populations at different interim analyses correspond to that of the subgroups
Group sequential type boundaries can be derived for z-tests, assuming that the variance is known. For each threshold qk, we define the z-statistic
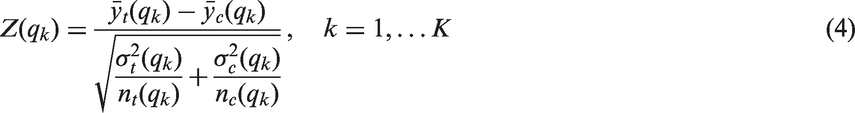
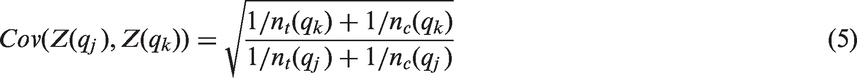

If Student’s t-tests to account for unknown variance instead of z-tests are applied at each stage, Jennison and Turnbull
8
propose to calculate the critical values as above (based on the multivariate normal distribution) and then to transform them to the corresponding boundary of the univariate t-distribution with

3.1.3 Multiple t-tests accounting for different variances across subgroups
Due to prognostic effects of the biomarker, the variances may vary across subgroups. Then the distributional assumptions on which the group sequential approach to calculate the critical boundaries is based on, are no longer met. However, the test statistics will still asymptotically follow a multivariate normal distribution, and under the global null hypothesis
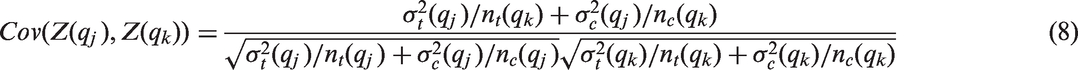
Assuming equal variances across treatment arms within a given subgroup this simplifies to
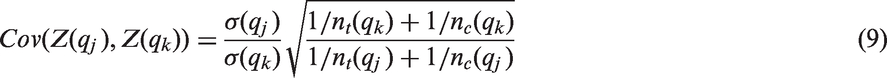
3.2 Regression models to adjust for prognostic biomarkers
An alternative approach to account for prognostic effects is a regression model for the treatment comparison. For example, adjusting for the biomarker as a covariate, we fit in each subpopulation
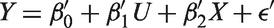
Then, for each subpopulation
The correlation structure of the test statistics can be approximated based on the group sequential approach by estimating the information for subgroup
Similar as for the group sequential t-test, the calculation of the critical boundaries relies on the assumption that the variance of the residuals is the same in all subpopulations. Thereby this approach extends the group sequential approach to the setting of prognostic biomarkers. While the assumption of a common variance across subpopulations hold if the fitted regression model is correct, the residual variances may vary across subpopulations if the model is misspecified.
3.3 Inverse normal combination tests
A multiple testing procedure for nested subpopulations can also be constructed using combination tests.10,13,15,16 To this end we split the population into disjoint subsets
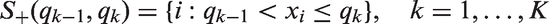
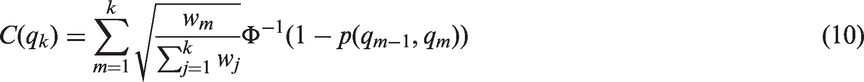
4 Properties of the multiple testing procedures
To investigate the operating characteristics of the procedures introduced in the previous section, a simulation study was performed. For simplicity, we assume the biomarker to be uniformly distributed on
We considered six testing procedures: Šidák adjusted t-tests, t-tests based on critical values
The data were generated based on the model given in equation (1) with per group sample sizes of n = 80. Simulation results for the FWER for n = 160 and 320 can be found in the Supplementary material.
We considered two scenarios. First, the step function model defined in equation (3) (see Figure 1(a)) with parameters
The second scenario considered is the linear trend model (see Figure 1(b)), where the prognostic and predictive effects of the biomarker on the outcome Y are linear. We considered settings where
In both scenarios, the variance of the noise term ε in equation (1) was set to 1.
4.1 Family-wise Type 1 error rate
The FWER for the considered procedures is shown in Figures 2 and 3 for the step function model and the linear trend model. If there is no prognostic effect, all considered methods control the FWER, with the exception of the z-test (which is only based on the normal approximation) which has an inflated error rate for the scenarios with small to moderate sample sizes.
FWER as a function of the prognostic effect assuming a step function dependence for a sample size of n = 80. The number of thresholds was set to K = 2, 4, 8 with true cut-off FWER and Power as a function of the prognostic (FWER-Plot) or predictive (Power-Plot) effect assuming a linear dependence for a sample size of n = 80. The number of thresholds was set to K = 2, 4, 8. The black lines show the results for the Šidák (solid), the z-test (FWER-Plot) or full population test (Power-Plot) (dot-dashed), the t-test (dotted) and the adjusted t-test (dashed) while the grey lines represent the regression procedure (dashed) and the inverse normal test (solid).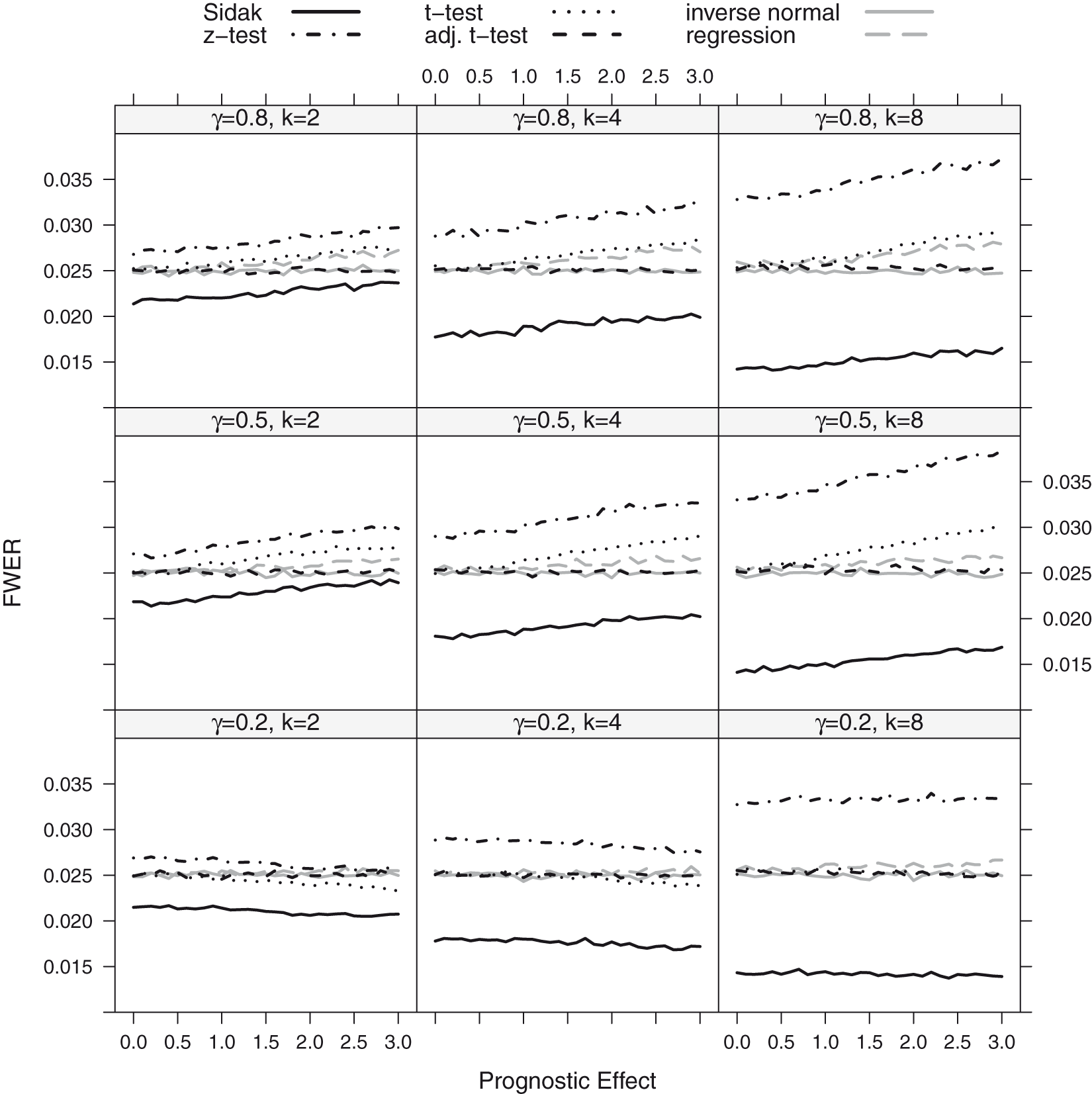
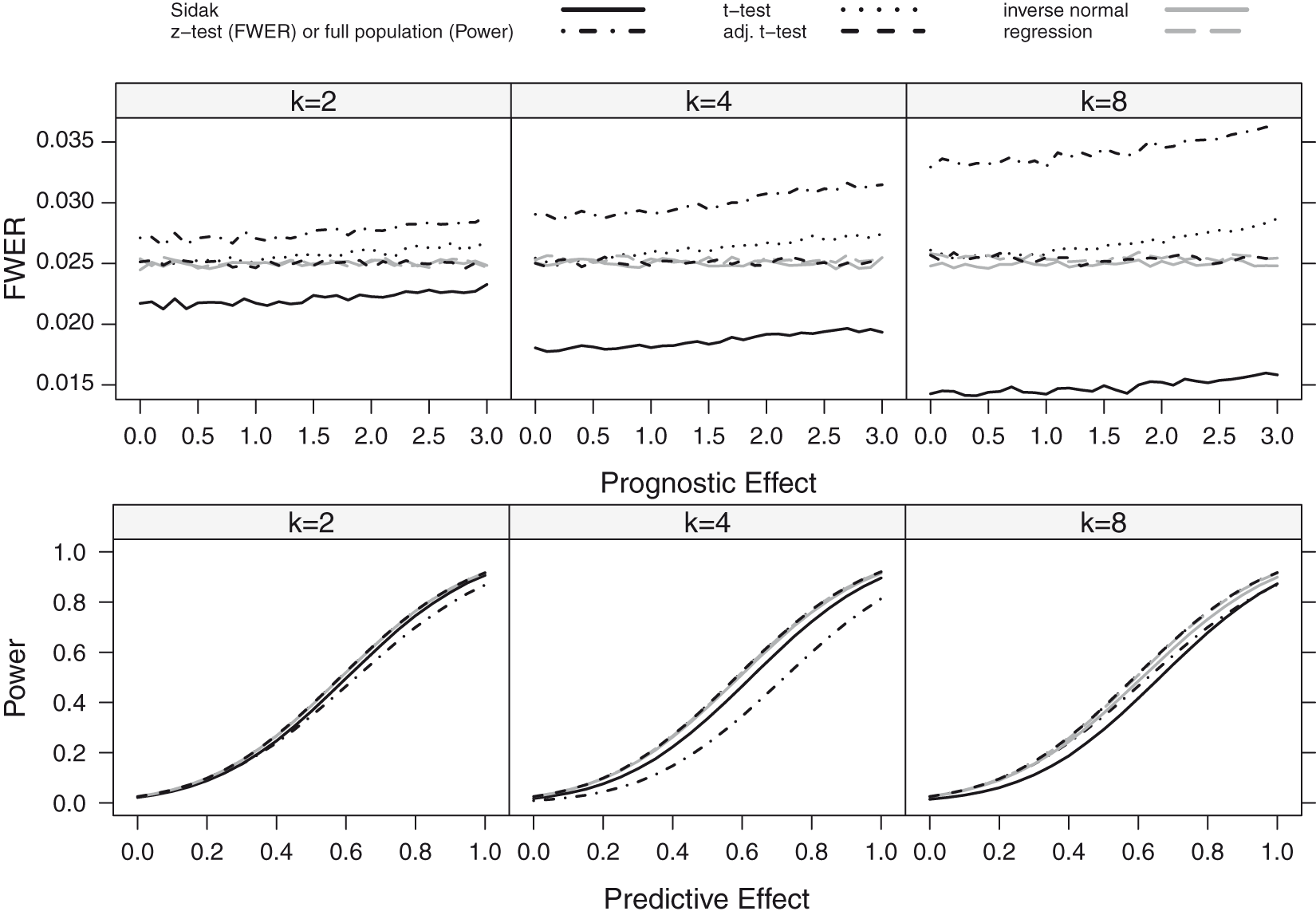
If the biomarker has, however, a prognostic effect, also the group sequential boundaries adjusted with t-quantiles (t-test) can become liberal. The amount of inflation depends on the size of the prognostic effect, the number of thresholds considered (the more thresholds, the larger the inflation) and the value of the true cut-off point γ. The observed inflation results from the effect of the prognostic effect on the variance of the outcome in the different subgroups. For larger prognostic effects, the variance of the outcomes in the different subgroups vary and this has an impact on the correlation structure between test statistics such that the assumptions underlying the computation of the critical boundaries based on a group sequential test are no longer satisfied. This leads in several settings to an inflation of the FWER when using group sequential boundaries. Note that (with the exception of the z-test with low or moderate sample sizes) substantial inflations of the FWER are only observed for prognostic effects larger than a standard deviation.
The regression procedure has a somewhat lower FWER for the step function model but remains anti-conservative because of the model misspecification. In the linear trend model, it controls the level well.
Across all scenarios, the t-test accounting for different subpopulation variances (adjusted t-test), the inverse normal combination test and the Sidák test control the FWER. However, the latter is strictly conservative, especially for a larger number of thresholds.
4.2 Power
We report the power of the procedures, defined as the probability to reject at least one of the K hypotheses. We did not consider the z-test in these simulations, as it did not sufficiently control the FWER for the considered sample size of n = 80. Instead, we also report the power of a single t-test in the full population, for comparison. The power for the step function and the linear trend model is shown in Figures 3 and 4. Under both scenarios, the approaches based on group sequential t-tests (t-test, adjusted t-test), the regression approach and the inverse normal combination test show similar power and the lines in the plot are partly indistinguishable. For K = 8 thresholds, the inverse normal combination test has a somewhat lower power compared to the group sequential t-test and the regression method because of a loss of degrees of freedom due to the split in disjoint subsets. Over all scenarios, the Šidák test shows the lowest power as it does not make full use of the correlation structure between the test statistics. If the size of the subpopulation is small ( Power as a function of the predictive effect assuming a step function dependence for a sample size of n = 80: the number of thresholds was set to K = 2, 4, 8 with true cut-off 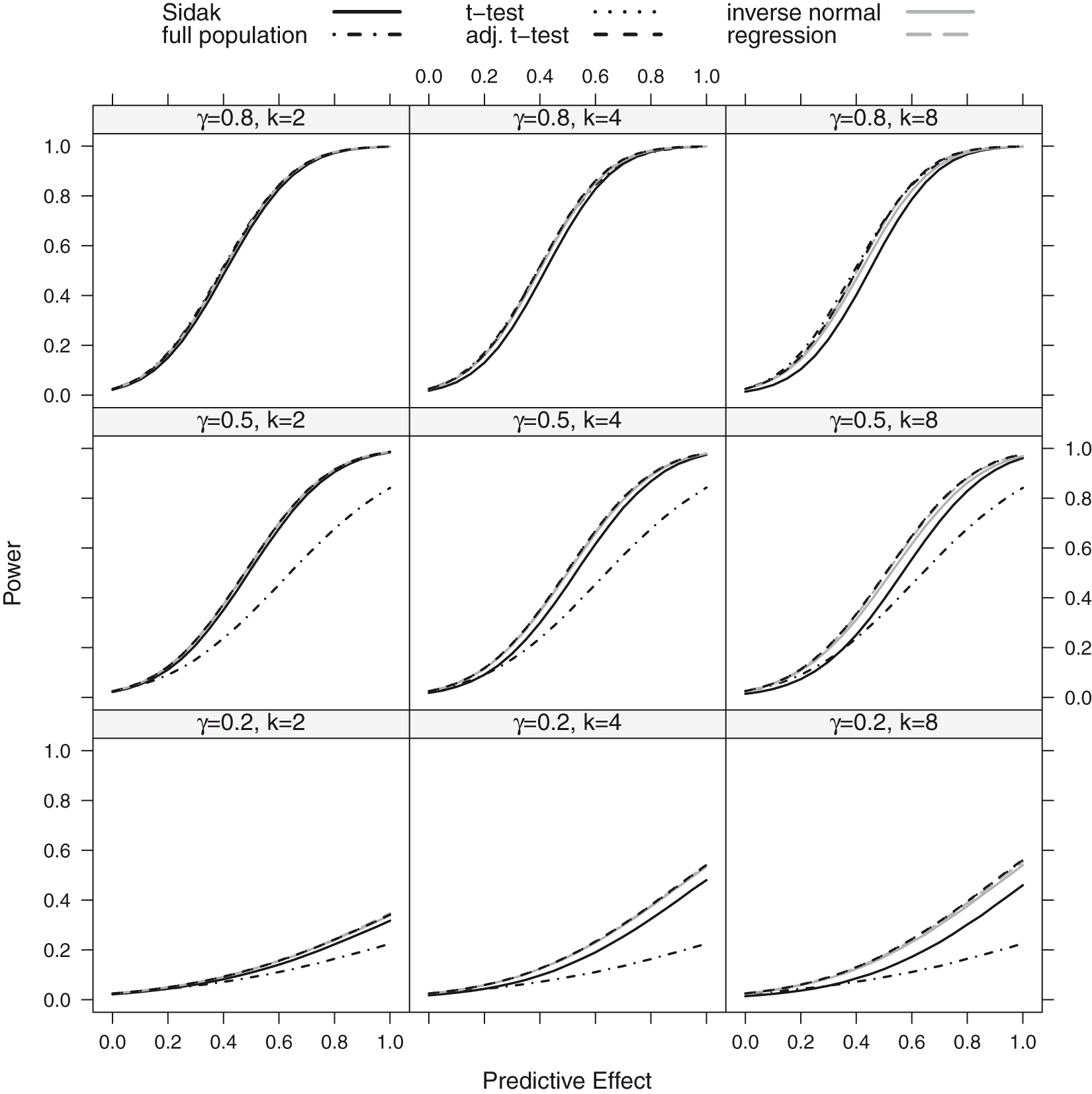
4.3 Model misspecifications
In the above simulations, the structure of the tested subgroups (where all patients with a biomarker value below a certain threshold are included) is in agreement with the considered scenarios, where the prognostic and predictive effects decrease monotonically with the biomarker (see Figure 1). To assess the robustness of the procedure, we investigated FWER and power if this assumption does not hold. As above, we assumed a step function model and a linear trend model but with monotonically increasing prognostic and predictive effects such that
5 Example: clinical trials in depression with a predictive biomarker
Depression is a common and disabling disease for which a number of pharmacological and psychosocial treatment options are available. However, no single treatment is universally effective and the response to treatment is slow and hard to predict. Therefore, many patients with depression undergo multiple treatments before achieving remission.1,2 One problem is the heterogeneity of the disease which has motivated the investigation of biomarkers to predict the treatment outcome. As outcome measures, in such studies often the decrease in a score describing the severity of the disease is used. Examples of commonly used instruments include the Montgomery-Asberg Depression Rating Scale (MADRS), the Hamilton Rating Scale for Depression (HRSD) or the Beck-Depressions-Inventar II Score (BDI-II).
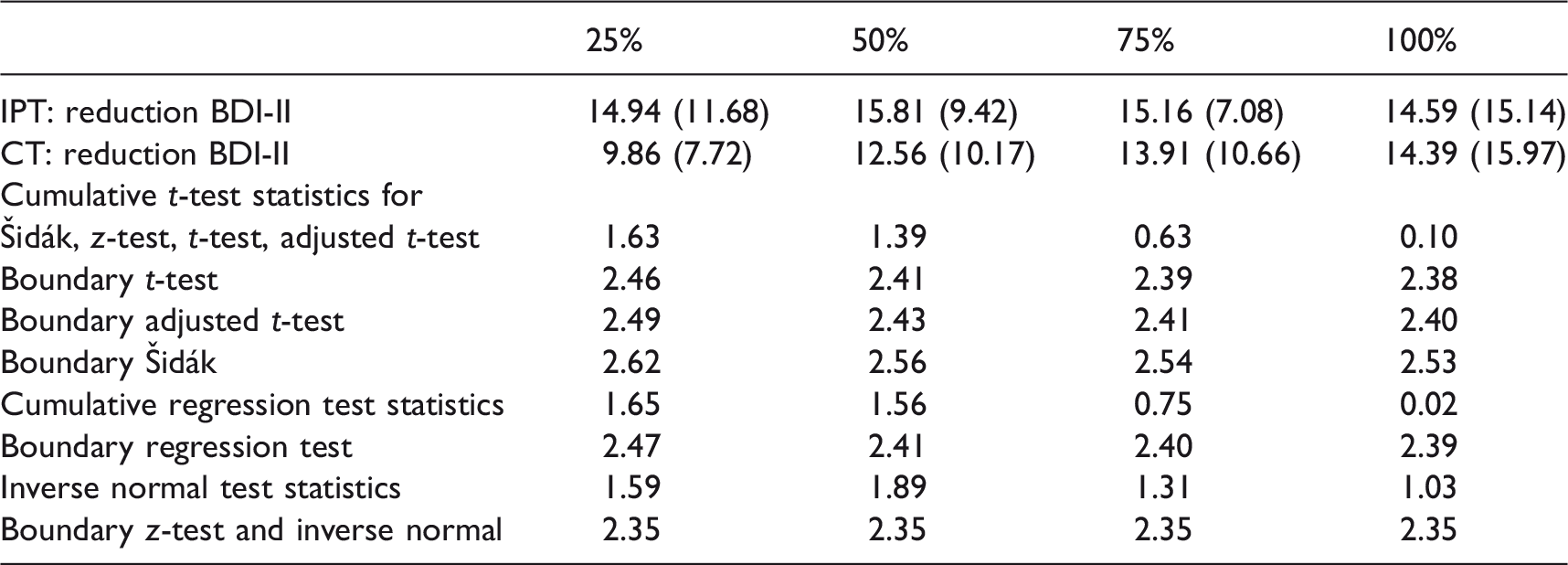
Luty et al. 18 compared in a randomized controlled trial interpersonal psychotherapy (IPT) and cognitive-behavioural therapy (CT) for major depression. A total of 177 patients were randomly allocated to the two treatment groups. As primary outcome variable, the percentage improvement in MADRS score from baseline to the end of a 16-week treatment phase was investigated. No statistically significant difference between IPT and CT was found for the full population. In a secondary analysis, however, investigators found that severely depressed patients responded significantly better to CT than to IPT, suggesting baseline severity as a predictor for response. To categorize severe depression, they used a fixed threshold for the baseline MADRS score. No correction for multiplicity was performed for the subgroup test.
Similarly, Lemmens et al. 19 compared IPT and CT in a randomized controlled trial also concluding that there is no statistically significant difference between the two treatments. The main outcome measure was the decrease in BDI-II score from baseline to seven months. Also 182 patients were randomized in three groups, 75 to IPT, 76 to CT and 31 patients were randomized to a waiting list control condition. Although no statistically significant difference between the two active treatment arms was observed, both treatments were superior to the waiting list group. In a re-analysis of the data based on the IPT and CT groups only, Huibers et al. 20 investigated several baseline scores (describing the severity of the disease), as, e.g., the Inventory of Interpersonal Problems Score (IIP), the Beck Hopeless Scale (BHS), the Brief Symptom Inventory (BSI) or quality of life scores as potential predictors for treatment outcome. Using a variable selection approach based on linear regression models with interaction terms, they found that, for example, the BSI Cognitive Problems score or the IIP self-sacrificing score may be moderators of treatment outcome.
Mean (SD) of the outcome, test statistics and critical boundaries of the Šidák, z-test, t-test and the corrected t-test as well as for the regression method and the inverse normal test separately for the four nested subgroups calculated from the example data set.
6 Multiple t-test for general subgroups
Consider a general set of subsets 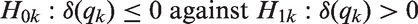
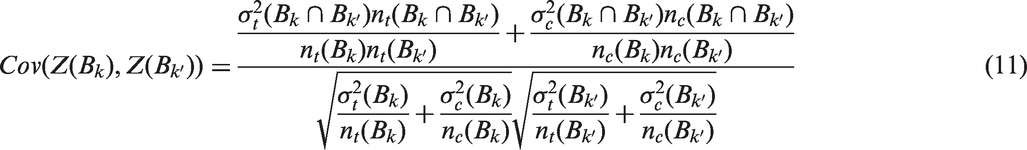
The normal approximation of the level α condition is given by
An application of this more general procedure are subgroups defined by the tail-oriented construction of the STEPP method,
5
which has been proposed in settings where there is uncertainty if very low or very large values of the biomarker are predictive for a large treatment effect. Here, first a left-to-right cumulation of patient values is performed where subgroups are defined by all subjects with biomarker values below a set of thresholds (as defined in the above sections) and then a right-to-left cumulation is performed where subgroups are defined by all subjects with biomarker values above the set of thresholds. Given K thresholds, this procedure defines
7 Discussion
We investigated methods for nested subpopulation tests, where the subpopulations are defined by thresholds of a continuous biomarker.
Our results show that special care has to be taken when using critical boundaries from group sequential designs, as has been proposed previously. If there are prognostic effects that are not adequately adjusted for, the standard critical boundaries from group sequential designs will not control the FWER in general. However, a substantial inflation of the FWER occurs for large prognostic effects only. Correcting for the least favorable correlation structure using the Šidák test controls the FWER. However, it can become very conservative if a larger number of subgroups are tested when it also leads to a loss in power. The inverse normal combination test controls the FWER but has slightly smaller power for a larger number of thresholds due to a loss in degrees of freedom.
Furthermore, the power calculations show that if the subpopulation, where the treatment effect is positive, is large, testing the null hypothesis for the full population only has similar power as compared to the multiple testing methods testing for a treatment effect in multiple subgroups. However, in settings where the subgroup where the treatment effect is positive, is smaller, the multiple tests have a larger power to reject at least one null hypothesis than the test for the full population only. The findings on the power imply that in these settings, the sample size required to achieve a certain power is lower for the multiple testing procedure than for the single test in the full population (accounting for the diluted treatment effect in the latter). The sample size yielding a certain power for the multiple testing procedures cannot be given explicitly, but can be obtained through numerical approximation or simulation techniques (see e.g. Placzek and Friede
6
). For example, assuming a true cut-off in the step function model of
Note that for the sample size calculation the thresholds must be chosen in the planning phase of a trial because the critical boundaries for the multiple t-test depend on the number of thresholds K as well as the size of the subgroups. If the subgroups are defined by absolute thresholds (rather than quantiles), the sample size calculation will be based on expected subgroup sizes since the actual subgroup sizes are random. In this case, at the final analysis the critical values need to be updated based on the actual subgroup sizes. Alternatively one may choose the thresholds based on quantiles of the continuous biomarker such that the subgroup sizes are fixed. This, however, results in data-dependent absolute thresholds.
In this manuscript, we focused on single-step multiple testing procedures. Using the closed testing principle, these can be improved by a sequentially rejective test. While this has no impact on the probability to reject at least one null hypothesis, it will increase the power to demonstrate a statistically significant treatment effect in several subgroups. Furthermore, for all the considered testing, multiplicity adjusted p-values can be defined by determining for each hypothesis the smallest significance level α, for which the test rejects the respective hypothesis.
The observed FWER inflation for group sequential tests of hypotheses for nested subpopulations has also implications for classical group sequential designs. A corresponding type 1 error rate inflation can occur also in group sequential tests of a single hypothesis if there is a time trend in the outcome variable. The calendar time then has a similar impact as the prognostic biomarker in the subpopulation tests and the classical group sequential test may have an inflated type 1 error rate.
An alternative approach to test for a treatment effect in nested subpopulations that has not been explored in this manuscript is to fit a single linear model including the factors treatment, as well as indicator functions of the disjoint sets
Supplemental Material
Supplemental material for Robustness of testing procedures for confirmatory subpopulation analyses based on a continuous biomarker
Supplemental Material for Robustness of testing procedures for confirmatory subpopulation analyses based on a continuous biomarker by Alexandra Christine Graf, Gernot Wassmer, Tim Friede, Roland Gerard Gera and Martin Posch in Statistical Methods in Medical Research
Footnotes
Acknowledgement
We thank the two reviewers for their useful comments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work has received funding from the European Unions 7th Framework Programme for research, technological development and demonstration under Grant Agreement no 602144 (InSPiRe).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.