
Abstract
Multi-arm multi-stage trials can improve the efficiency of the drug development process when multiple new treatments are available for testing. A group-sequential approach can be used in order to design multi-arm multi-stage trials, using an extension to Dunnett’s multiple-testing procedure. The actual sample size used in such a trial is a random variable that has high variability. This can cause problems when applying for funding as the cost will also be generally highly variable. This motivates a type of design that provides the efficiency advantages of a group-sequential multi-arm multi-stage design, but has a fixed sample size. One such design is the two-stage drop-the-losers design, in which a number of experimental treatments, and a control treatment, are assessed at a prescheduled interim analysis. The best-performing experimental treatment and the control treatment then continue to a second stage. In this paper, we discuss extending this design to have more than two stages, which is shown to considerably reduce the sample size required. We also compare the resulting sample size requirements to the sample size distribution of analogous group-sequential multi-arm multi-stage designs. The sample size required for a multi-stage drop-the-losers design is usually higher than, but close to, the median sample size of a group-sequential multi-arm multi-stage trial. In many practical scenarios, the disadvantage of a slight loss in average efficiency would be overcome by the huge advantage of a fixed sample size. We assess the impact of delay between recruitment and assessment as well as unknown variance on the drop-the-losers designs.
Keywords
1 Introduction
Testing multiple experimental treatments against a control treatment in the same trial provides several advantages over doing so in separate trials. The main advantage is a reduced sample size due to a shared control group being used instead of a separate control group for each treatment. Other advantages include that direct comparisons can be made between experimental treatments and that it is administratively easier to apply for and run one multi-arm clinical trial compared to several traditional trials. 1 Multi-arm multi-stage (MAMS) clinical trials include interim analyses so that experimental treatments can be dropped if they are ineffective; also, if desired, the trial can be designed so that it allows early stopping for efficacy if an effective experimental treatment is found. Two current MAMS trials that are ongoing are the MRC STAMPEDE trial, 1 and the TelmisArtan and InsuLin Resistance in HIV (TAILoR) trial (the design of which is discussed in Magirr, Jaki and Whitehead 2 ).
Magirr et al. 2 extend Dunnett’s multiple-testing procedure 3 to multiple stages, which we refer to as the group-sequential MAMS design. In this design, futility and efficacy boundaries are prespecified for each stage of the trial. At each interim analysis, statistics comparing each experimental treatment to the control treatment are calculated and compared to these boundaries. If a statistic is below the futility boundary, then the respective experimental arm is dropped from the trial. If a statistic is above the efficacy threshold, the trial is stopped with that experimental treatment recommended. Boundaries would generally be required to control the frequentist operating characteristics of the trial. Since there are infinitely many boundaries that do so, a specific boundary can be chosen to minimise the expected number of recruited patients at some treatment effect, 4 or by using some boundary function such as those of Pocock, 5 O’Brien and Fleming, 6 or Whitehead and Stratton. 7
The group-sequential MAMS design is efficient in terms of the expected sample size recruited, but has the practical problem that the sample size used is a random variable. This makes planning a trial more difficult than when the sample size is known in advance. An academic investigator applying for funding to conduct a MAMS trial will find that traditional funding mechanisms lack the required flexibility to account for a random sample size. 8 Generally, they would have to apply for the maximum amount that could potentially be used, with the consequence that such trials appear highly expensive to fund. There are also several other logistical issues to consider, such as employing trial staff to work on a trial with a random duration.
An alternative type of MAMS trial is one in which a fixed number of treatments is dropped at each interim analysis. Stallard and Friede 9 propose a group-sequential design where a set number of treatments is dropped at each interim analysis, and the trial stops if the best-performing test statistic is above a predefined efficacy threshold or below a predefined futility threshold. The stopping boundaries are set assuming the maximum test statistic is the sum of the maximum independent increments in the test statistic at each stage, which is generally not true and leads to conservative operating characteristics. A special case of Stallard and Friede’s design is the well-studied two-stage drop-the-losers design,10,11 in which one interim analysis is conducted, and only the top-performing experimental treatment and a control treatment proceed to the second stage. In Thall et al., 10 the chosen experimental treatment must be sufficiently effective to continue to the second stage. More flexible two-stage designs have been proposed by several authors, including Bretz et al. 12 and Schmidli et al. 13 These designs used closed testing procedures and/or combination tests to control the probability of making a type-I error whilst allowing many modifications to be made at the interim. In the case of multiple experimental arms, there is more scope for improved efficiency by including additional interim analyses, at least for group-sequential MAMS designs.2,4
In this paper, we extend the two-stage drop-the-losers design to more than two stages and derive formulae for the frequentist operating characteristics of the design. The resulting design has the advantage of a fixed sample size by maintaining a prespecified schedule of when treatments are dropped. That is, at each interim analysis, a fixed number of treatments are dropped. Note that this could be thought of as subdividing the first stage of a two-stage drop-the-losers trial to allow multiple stages of selection. We show that when there are several treatments, allowing an additional stage of selection noticeably decreases the sample size required for a given power, compared to the two-stage design. We also compare the multi-stage drop-the-losers design to the Dunnett-type MAMS design.
2 Notation
We assume that the trial is to have J stages, that is, J − 1 interim analyses and a final analysis, and starts with K experimental treatments and a control treatment. Let
For
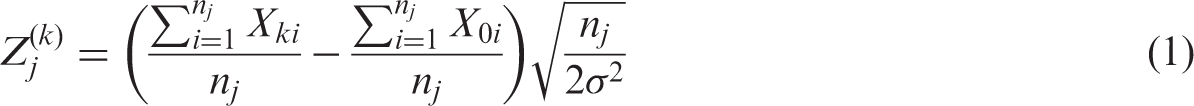
The covariance between different test statistics can be shown to be
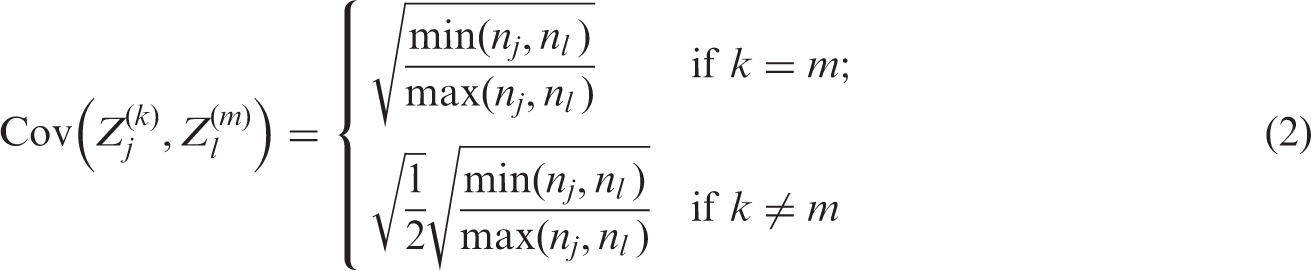
It is desirable that the design is chosen in order to control the family-wise type-I error rate (FWER). The FWER is the probability of rejecting at least one true null hypothesis, and strong control of the FWER at level α means that the FWER is
3 Analytic operating characteristics
In this section, we provide analytical formulae for the probability of a particular treatment being recommended under a general vector of treatment effects. We also provide formulae for the probability of rejecting any null hypothesis when HG is true, and the probability to select the best treatment under the LFC. Although the formulae extend naturally to more than three stages, the expressions grow in length with the number of stages. For simplicity of exposition, we concentrate on the three-stage case, where K experimental treatments are included in the first stage, L < K in the second stage, and 1 in the third stage. This is denoted as the
3.1 Probability of a specific treatment being recommended
For subsequent development, it is useful to define a ranking of the experimental treatments in terms of how successful they are in the trial. We introduce random variables the treatment that reaches the final analysis has rank 1; the treatment that is dropped at the first analysis with the lowest test statistic is given rank, K; if treatment k1 reaches a later stage than treatment k2, then if treatments k1 and k2 are dropped at the same stage, and k1 has a higher test statistic at that stage, then
For instance, for a three-stage 4:2:1 design where treatment 3 reaches the final stage, treatment 2 is dropped at the second analysis, treatments 1 and 4 are dropped at the first analysis, and treatment 1 has the lowest test statistic at the first analysis, the realised value of ψ is (4, 2, 1, 3).
For J = 3, the probability of recommending treatment k, that is, rejecting
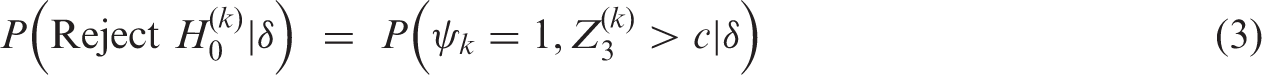

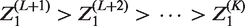
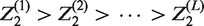
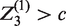



Other terms in equation (4), in which the values of
The above approach extends directly to designs with more than three stages. For a
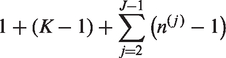
3.2 Probability of recommending any treatment under the global null hypothesis
When the global null hypothesis HG is true, each element of m(δ) is 0. By symmetry, the probability of observing each ordering ψ and a final Z statistic greater than c is the same. Thus, the probability of recommending any treatment under the global null hypothesis is
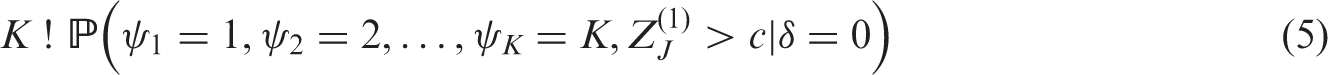
3.3 Probability of recommending a specific treatment under the LFC
We assume the trial is to be powered to recommend treatment 1 at the LFC, where
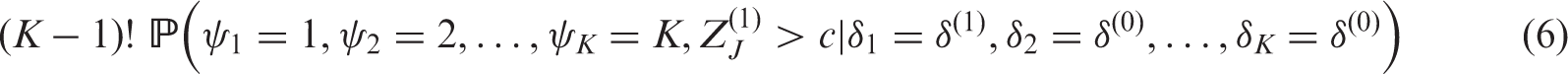
R code provided online (https://sites.google.com/site/jmswason) allows the user to find the values of n and c so that a design has required FWER and power.
4 Strong control of FWER
We can control the probability of recommending an ineffective treatment when the global null hypothesis HG is true by specifying the critical value c so that the probability (5) is equal to α. In the case of a group-sequential MAMS trial, controlling the error rate under HG has been shown to control the FWER in the strong sense. 2 In this section, we prove that controlling the FWER at the global null hypothesis strongly controls the FWER for the multi-stage drop-the-losers design also.
We denote by mj, the fixed number of observations collected in stage j on each surviving treatment and on the control arm. At the end of stage j, the cumulative sample size on each remaining treatment and the control arm is
Initially the set of indices of all treatments is
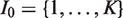
Recall for
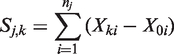
We first consider the general case where treatments 1 to K have treatment effects
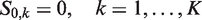
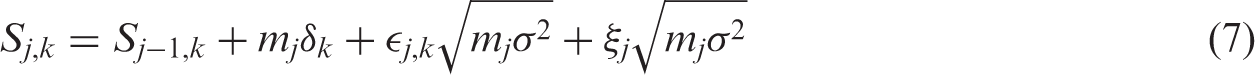
After the data-gathering part of stage j, the treatment
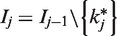

The trial is designed to have type-I error probability α when
Consider two trials that have the same design but differ with respect to values of the treatment effects. In Trial 1,
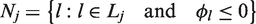
With
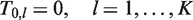
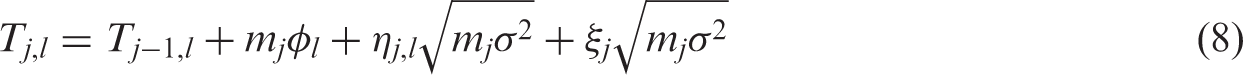
After the data-gathering part of stage j, the treatment
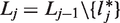

We shall establish the desired FWER property by a coupling argument, which assumes the terms ξj in equations (7) and (8) are equal and which reuses values ηj,
l
in equation (8) as values for some of the
A key step in the coupling argument is to define the relationship between treatments
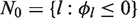
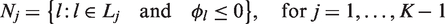
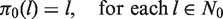
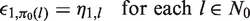


Assuming we can define the desired functions πj, there are two possibilities at the end of the trial when stage j = K is completed. The first possibility is that, on entering stage K, the set
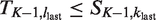
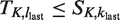

It remains to show that injective functions πj from Nj to Ij,

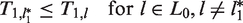
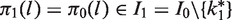
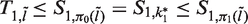
The construction of functions πj for
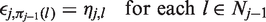
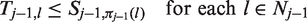
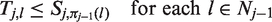
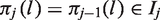
As noted earlier, if
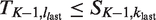
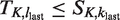

5 Results
5.1 Motivating trial
As a case study for the results in this paper, we consider the currently ongoing TAILoR trial, the design of which is discussed in Magirr et al. 2 This trial was originally designed to test four different doses of Telmisartan. Telmisartan is thought to reduce insulin resistance in HIV-positive individuals on combination antiretroviral therapy. The primary end point was reduction in insulin resistance in the telmisartan-treated groups in comparison with the control group as measured by homeostatic model assessment – insulin resistance (HOMA-IR) at 24 weeks. A group-sequential MAMS design was used to avoid assumptions regarding monotonicity of dose–response relationship, which were thought to be invalid based on a previous trial of the treatment in a different indication.
The trial design controls the FWER at 0.05 with 90% power under the LFC with
5.2 Comparison of two- and three-stage drop-the-losers designs
We first show that extending the drop-the-losers design beyond two stages can be worthwhile. For
Sample sizes required for a one-stage design and two-stage and three-stage drop-the-losers designs with α = 0.05, β = 0.1, δ(1) = 0.545 and δ(0) = 0.178.
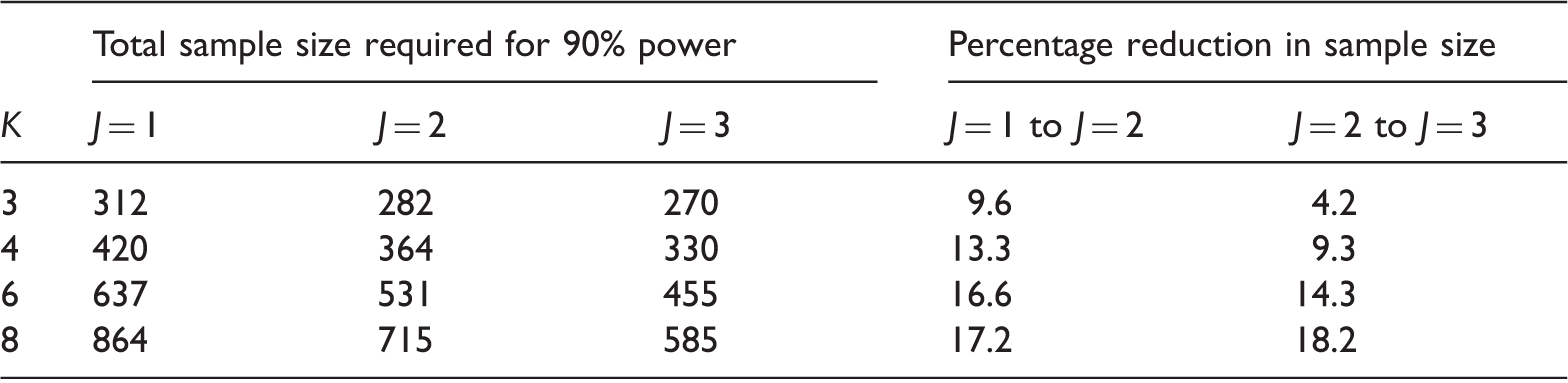
Note: For each three-stage design, the number of treatments proceeding to stage 2 is chosen to give the lowest total sample size: in the notation of Section 2, these designs are 3:2:1 for K = 3, 4:2:1 for K = 4, 6:3:1 for K = 6 and 8:3:1 for K = 8.
5.3 Comparison of three-stage group-sequential MAMS and drop-the-losers designs
We now compare sample size properties of drop-the-losers designs with those of group-sequential MAMS designs when design parameters are specified as in the previous section. The group-sequential MAMS designs have three analyses and use the triangular test boundaries of Whitehead and Stratton,
7
which are known to give good expected sample size properties.
4
Figure 1 shows boxplots of the sample size distribution (using 250,000 replicates) for the three-stage group-sequential MAMS designs with K = 4 and K = 6 experimental arms under four scenarios: (1) under HG; (2) under the LFC; (3) when δ1 = δ2 =… = δK = δ(0) and (4) when δ1 = δ2 =… = δK = −δ(0). The solid black line in each boxplot represents the median sample size. The dashed line for each K represents the fixed sample size of the most efficient three-stage drop-the-losers designs ( Sample size distribution for three-stage group-sequential MAMS designs with K = 4 and K = 6 and four vectors of treatment effects. Scenario 1 – the global null hypothesis (HG); scenario 2 – the LFC; Scenario 3 – all experimental treatments have uninteresting treatment effect δ(0); Scenario 4 – all experimental treatments have effect 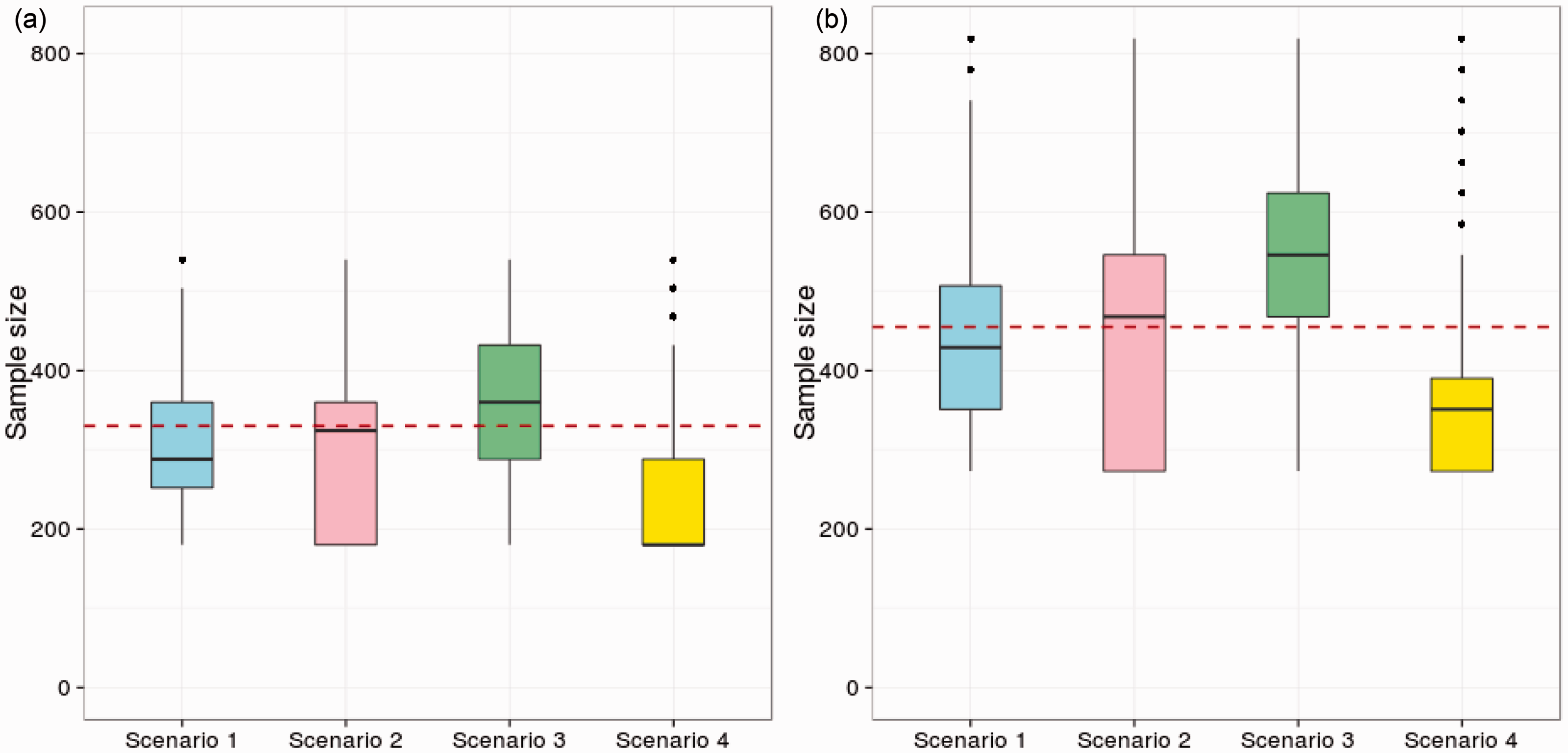
Although the group-sequential MAMS designs with triangular test boundaries are known to have low expected sample sizes, Figure 1 shows that the sample size distribution is highly variable and depends strongly on the configuration of treatment effects. If we take the median sample size of the group-sequential MAMS design as a point of comparison, we see the sample size for the drop-the-losers design is higher under HG (Scenario 1), almost equal under the LFC (Scenario 2) and lower when all treatment effects are equal to δ(0) (Scenario 3). These results are generally encouraging for the drop-the-losers design and show that the constraint of a fixed total sample size can be met without sacrificing much efficiency in terms of average numbers of patients recruited.
The performance of the drop-the-losers design is poorest in Scenario 4 where all the treatment effects are negative and the MAMS designs are likely to stop the whole trial early for futility. Results for this scenario indicate the desirability of adding a futility rule to the drop-the-losers design: although some variation in total sample size would be introduced, ethical considerations argue against continued use of treatments which are proving ineffective. One might, for example, specify a minimum requirement for treatments to meet at each stage and allow fewer than the specified number to continue when some treatments fail to meet this requirement – or stop the trial completely if no treatment satisfies the requirement. If a rule of this type was superimposed on the drop-the-losers design with no other changes to sample numbers or the final critical value, c, the type-I error rate would simply be reduced. Alternatively, the calculations of Section 3.1 could be extended to include this form of futility rule and the design parameters adjusted to satisfy the type-I error rate requirement exactly.
6 Spacing of interim analyses when there is delay between recruitment and assessment of patients
In previous sections, we have assumed there is no delay between recruitment and assessment of patients. In reality, there will nearly always be some delay, and often it will be considerable. For example, in the TAILoR trial, the final end point is measured 24 weeks after treatment.
A delay between recruitment and assessment means that at the time of an interim analysis, there will be patients who have been recruited but not yet assessed, and thus contribute no information to that interim analysis. The efficiency of the trial, in terms of number of patients recruited, is then reduced as some patients will be recruited to arms that are dropped before their responses are measured. Also, with a delay in response there are fewer observations at each interim analysis and, thus, lower probabilities of selecting the best treatments. The potential loss of efficiency depends on the recruitment rate to the trial since this rate and the time at which the final end point is measured together determine the numbers of patients treated but not assessed at the interim analyses.
Hampson and Jennison 15 have proposed ways of using partial information from patients who have been recruited but not assessed at the time of an interim analysis. If a short-term end point that is correlated with the final end point is available, fitting a joint model for both end points can increase the information for the final end point. When the final end point is the incidence of an event before a certain time, t* say, inference can be based on a Kaplan–Meier estimate of the probability of the event occurring before t*. In this case, the time-to-event data for all patients is used, with right censoring applying when the follow-up time is less than t* and the event has not yet occurred.
When there is a delay in response, the methodology described in Sections 3.1–3.3 can still be applied by conducting analyses at times when the required numbers of observations become available. We have explored the optimal spacing of analyses when there is a known delay. Since we have efficient computational methods for drop-the-losers designs, it is quite feasible to explore a wide variety of spacings. We report results for an example in which the primary end point is measured 24 weeks after recruitment, as in the TAILoR trial, and we consider recruitment rates of m = 1, 2 and 4 patients per week. The limiting case m = 0 is also included to represent the case of an immediate response.
We consider the 4:2:1 and 4:1 designs with, as before, δ(1) = 0.545, δ(0) = 0.178, α = 0.05 and
Properties of 4:2:1 and 4:1 designs when there is a 24-week delay between recruitment and assessment.

Note: A constant recruitment rate of m patients per week is assumed. Here, SS denotes sample size and m = 0 represents the limiting case when there is no delay in observing the response.
Table 2 shows that as the recruitment rate increases, there is a lower efficiency gain from including interim analyses. With a single interim analysis, the reduction in sample size of 14% in the case of immediate response falls to 7.1% when m = 2 and is lost completely for m = 4. The advantage of a three-stage design over a two-stage design also falls as m increases. Optimising the timing of the interim analyses is important here. As an example, with m = 2, a 4:2:1 design with equally spaced interim analyses, that is, (ω2, ω3) = (1, 1), needs a total of 390 patients, compared to the 363 patients for a design with the optimal spacing.
In view of these results, it is advisable to assess the likely impact of a delay in response on the efficiency of an adaptive design. Nevertheless, we have still seen that, for plausible combinations of recruitment rate and time to response, including either one or two interim analyses can reduce the sample size requirement compared to a design without interim analyses.
7 Discussion
MAMS designs are of great interest in practice, as their use means more new treatments can be tested with the same limited pool of patients. Much of the methodology about designing MAMS trials has focused on designs in which treatments are dropped early if their test statistics are below some prespecified futility boundary. This leads to variability in the number of treatments that will be in the trial at each stage, and therefore uncertainty in the total sample size required. This leads to uncertainties in applying for funding to conduct a MAMS trial, as well as other logistical issues such as staff employment. A design that does have a fixed sample size is the two-stage drop-the-losers design, where multiple experimental treatments are evaluated at an interim analysis, then the best-performing experimental treatment goes through to the second stage. We have investigated design issues in extending the drop-the-losers design to have more than two stages. If there are four or more treatments, we find that a third stage results in a considerable reduction in sample size. In addition, the fixed sample size compares well to the median sample size used in a group-sequential MAMS design. The design therefore retains many of the efficiency benefits of a MAMS design whilst also having a fixed sample size, which is very useful in practice. We have mainly considered the utility of adding a third stage, as each additional interim analysis increases the administrative burden of the trial. Adding a fourth stage provides a substantially lower additional efficiency advantage unless there are a lot of treatments being tested.
In this paper, we assumed a known variance of the normally distributed outcome. However, the method of quantile substitution, described in Section 3.8 of Jennison and Turnbull, 16 can be used to change the final critical value so that the type-I error rate is controlled when the variance is estimated from the data. We carried out simulations that showed this method performs very well in practice (results not shown), similarly to the group-sequential 17 and group-sequential MAMS cases. 4
In practice, the requirement to drop a fixed number of treatments at each stage may be difficult to keep to. For example, if all treatments are performing poorly in comparison to control, then it may be unethical to continue with even the best performing treatment. Any changes to the design during the trial will affect the operating characteristics of the trial. However, dropping more treatments than planned will lead to a lower than nominal FWER rather than an inflation. If one wishes to keep more treatments in the trial than originally planned, then this will lead to an inflation in FWER. However, by modifying the final critical value suitably, this inflation can be reduced. The analytical formulae in this paper can be modified in order to calculate the required critical value if more sophisticated stopping rules are used.
An alternative design that controls the number of treatments passing each analysis but also allows early stopping of the trial for futility or efficacy is the design of Stallard and Friede. 9 The multi-stage drop-the-losers design is somewhat less flexible than the Stallard and Friede design, but does have the advantage of having analytical formulae that provide exact operating characteristics of the design. The formulae for the Stallard and Friede design are conservative, especially when there are more than two stages. Of course simulation could be used to evaluate the operating characteristics exactly, but this makes it difficult to evaluate a large number of potential designs. We have shown that this is important in the case of delay between recruitment and assessment, where the spacing of the interim analyses becomes very important. The multi-stage drop-the-losers design can be evaluated extremely quickly, which allows the optimal interim analysis spacing to be found.
One worrying factor for the efficiency of adaptive trials in general, and the drop-the-losers design specifically, is delay between recruiting a patient and assessing their outcome. Such delay means that at a given interim analysis, there will be patients who are recruited but not yet assessed. These patients will not contribute to that interim analysis or to any subsequent analysis if the treatment they are on is dropped. We have investigated the effect of delay and show that drop-the-losers designs can still provide efficiency gains over a multi-arm design without interim analyses if the recruitment rate is below some level. This level will depend on the extent of delay and the total sample size of the trial. There are two factors that may go someway towards mitigating the impact of delay. Firstly, there may well be early outcomes that correlate well with the final outcome. 18 For example, in the TAILoR trial, the final outcome is HOMA-IR at 24 weeks, but if earlier measurements could be made, these may well be highly informative for the 24 week end point. In that case, more patients could be included in the interim analysis. A second factor is that trial recruitment tends to start slowly and increase over time, perhaps as more centres are added to the trial. This means that a greater proportion of patients may be available for assessment at earlier interim analyses compared to the uniform recruitment case we considered here. Research into the effect of delay on group-sequential MAMS trials and strategies to account for it (extending the work of Hampson and Jennison 15 to multi-arm trials) would be very useful.
This paper has considered design issues in multi-stage drop-the-losers trials. A drawback of adaptive designs in general is that estimation of relevant quantities, such as the mean treatment effect, after the trial is more complicated than in a traditional trial. For example, using the maximum likelihood estimate in two-stage trials will result in bias.19,20,21 The issue of estimation for multi-stage drop-the-losers trials is considered in Bowden and Glimm. 22
Footnotes
Acknowledgements
We thank Dr Ekkehard Glimm and two anonymous referees for their useful comments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the Medical Research Council (grant numbers G0800860 and MR/J004979/1).