
Abstract
Objective
To evaluate the long-term efficacy and safety of different doses of BI 655064 versus placebo added to standard of care during maintenance treatment for lupus nephritis (LN).
Methods
1293.13 was an exploratory, phase II maintenance trial. Patients were eligible for entry if they had completed 1 year of randomised treatment with BI 655064 (120, 180 or 240 mg) or placebo in the 1293.10 trial, responded to treatment at Year 1 (complete renal response [CRR], partial renal response or urinary protein/creatinine ratio ≤1) and consented to continue treatment. The primary endpoint was the proportion of patients with CRR without renal flares at Year 2. Secondary endpoints included change from baseline in Safety of Estrogens in Lupus Erythematosus National Assessment-Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI) scores and safety/tolerability.
Results
69/121 patients (57.0%) from the 1293.10 trial entered 1293.13. The adjusted proportion of patients with CRR decreased in all groups between Year 1 (BI 655064: 53.4%–72.7%; placebo: 71.4%) and Year 2 (BI 655064: 48.2%–59.5%; placebo: 57.5%). At Year 2, mean decreases in total SELENA-SLEDAI scores were greatest with BI 655064 240 mg (−10.6 points), followed by 120 mg (−8.9 points), 180 mg (−7.2 points) and placebo (−5.3 points). SELENA-SLEDAI non-renal scores decreased at Year 1 (BI 655064: −3.0 to −3.4; placebo: −1.8); this pattern remained with BI 655064 during Year 2 (−2.4 to −4.1), whereas placebo returned to near-baseline scores (−0.4). Over 2 years of treatment, almost all patients (97.1%) experienced ≥1 adverse event (AE). Compared with the other groups, higher rates of serious AEs (42.9% vs 23.1%–33.3%)—mainly driven by serious infections (23.8% vs 7.7%–14.3%)—and severe AEs (47.6% vs 13.3%–28.6%) were reported with BI 655064 240 mg.
Conclusions
This exploratory, phase II maintenance trial failed to demonstrate the benefits of BI 655064 on renal outcomes in the treatment of LN. However, some benefits in total and non-renal SELENA-SLEDAI scores were observed.
Introduction
Lupus nephritis (LN) is a serious complication of systemic lupus erythematosus (SLE) that occurs in 40%–60% of individuals.1–3 The standard of care (SoC) for LN (class III–IV) comprises cyclophosphamide or mycophenolate mofetil (MMF) with glucocorticoids, with the option of azathioprine (AZA) in patients with a response after 6 months.2–4 However, efficacy with SoC is limited, with up to two-thirds of patients not reaching complete renal response (CRR).5,6
Although recently approved treatments (voclosporin or belimumab—both as add-on to SoC) have improved responses,7,8 the need for novel treatment options remains. Interaction between CD40 and CD40 ligand (CD40L), both in the renal interstitium and elsewhere, is critical for generating autoantibody-producing B cells in LN, resulting in renal injury.1,9,10 Therefore, targeting the CD40–CD40L pathway is a promising approach in the treatment of LN.1,11
BI 655064, a second-generation humanised anti-CD40 antibody, has fragment crystallisable (Fc) regions with two mutations that prevent Fc-mediated antibody-dependent or complement-mediated cellular cytotoxicity and platelet activation.1,12,13 An exploratory, randomised, placebo-controlled, proof-of-concept, dose-finding trial (1293.10; NCT02770170) failed to demonstrate a dose–response relationship for the primary endpoint in patients with active LN following 1 year of treatment with BI 655064 as add-on to SoC (MMF/AZA plus glucocorticoids); the proportions of patients with CRR were similar across the active and placebo treatment groups (38.3%–48.3%). 14 However, potential benefits of BI 655064 on non-renal endpoints were demonstrated, including Safety of Estrogens in Lupus Erythematosus National Assessment-Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI), Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F) and the Lupus Patient-Reported Outcome tool (LupusPRO). Although most patients (86%–98% across all treatment groups including placebo) experienced adverse events (AEs), with 60%–75% of patients experiencing AEs classed as infections and infestations, BI 655064 was generally well tolerated. 14 Patients who had a meaningful renal response after 1 year of treatment in the 1293.10 induction trial could continue treatment for a second year in the 1293.13 maintenance trial. 15
Here, we report the efficacy and safety data after 2 years of BI 655064 treatment versus placebo (as add-on to SoC).
Methods
Study design
1293.13 was an exploratory, non-randomised, phase II maintenance trial (Figure S1) designed to evaluate the long-term efficacy and safety of different doses of BI 655064 versus placebo (as add-on to SoC), and to evaluate the effects of glucocorticoid tapering during the second year of treatment (Supplemental Appendix S1).
Treatment
Patients in the 1293.13 maintenance trial continued their regimens from the 1293.10 induction trial (which used a 2:1:1:2 randomisation for 1 year of treatment 14 ), with weely subcutaneous injections of placebo or BI 655064 (120, 180 or 240 mg; the full dose achieved over 2 weeks) for another year of therapy. All patients continued to receive SoC, comprising either MMF (1–2 g/day) or AZA (2 mg/kg/day) in combination with tapering doses of glucocorticoids (≤10 mg/day) (Supplemental Appendix S1 and Table S1). Withdrawal of BI 655064 or placebo did not necessarily lead to withdrawal from the trial, and the reasons for withdrawal from therapy or the trial were recorded. For patients who terminated their therapy early, an end-of-treatment visit occurred within 7 days of discontinuing therapy, and patients were encouraged to attend follow-up visits.
Patients
Eligible patients had completed 1 year of treatment in the induction trial, had responded to treatment at Year 1 (CRR, partial renal response [PRR], or proteinuria ≤1.0 g/day or urinary protein/creatinine ratio [UP/UC] ≤1) and consented to continue treatment in the maintenance trial. Full exclusion criteria are listed in Supplemental Appendix S1.
Written informed consent was provided by each patient. The study was conducted and reported in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and other applicable regulatory requirements, which were reviewed and approved by the Independent Ethics Committees and/or Institutional Review Boards of participating sites.
Endpoints
The primary endpoint was the proportion of patients with CRR without renal flares during Year 2. CRR was defined as UP <0.5 g/day (calculated as the mean UP from two 24-hour urine collections) and either estimated glomerular filtration rate (eGFR) ≥90 mL/min/1.73 m2 or, if the eGFR was <90 mL/min/1.73 m2 at the time of assessment, any decrease in eGFR had to be less than 20% from the baseline value. Renal flares were defined relative to proteinuria or eGFR at baseline. If proteinuria was ≤0.5 g/day at baseline, renal flare was defined as any unexplained increase (e.g. not due to infection or injury) in proteinuria to >1 g/day (or UP/UC >1) during the treatment period, confirmed by a repeat test within 2 weeks. If proteinuria was >0.5 g/day at baseline, and there was an initial decrease (≤0.5 g/day [or UP/UC ≤0.5] at ≥2 consecutive visits), renal flare was defined as any unexplained increase in proteinuria (>1 g/day, or UP/UC >1) confirmed by a repeat test within 2 weeks. If proteinuria was >0.5 g/day at baseline and there was no initial decrease, renal flare was defined as >2 times baseline (or UP/UC >2 times baseline), confirmed by a repeat test within 2 weeks. Renal flares were also defined as any decrease in eGFR by >20% from baseline and eGFR <90 mL/min/1.73 m2 (confirmed by a repeat test within 2 weeks) if worsening of eGFR was unexplained.
Secondary endpoints at Year 2 included the proportion of patients with confirmed CRR (cCRR; CRR at Weeks 42 and 52, using spot urine UP/UC ratios) without any renal flares at Week 52; CRR (UP 24-hour collection) without renal flares in patients who had sustained prednisone (or equivalent) reduction ≤5 mg from Week 26 of the maintenance trial; and PRR without renal flares (UP 24-hour collection at Week 52). Changes from baseline in SELENA-SLEDAI total, renal and non-renal scores, and in individual domains and without serological variables at Weeks 12, 26, 42 and 52 during Year 2, were also evaluated. Additional endpoints are described in Supplemental Appendix S1, as well as pharmacokinetics and immunogenicity analyses.
Safety
The safety of BI 655064 was assessed by evaluating AEs and tolerability over 2 years of treatment. The severity of AEs was graded according to the Rheumatology Common Toxicity Criteria v2.0. 16
Statistical methods
Analyses of the primary and secondary endpoints, which were exploratory and descriptive in nature, were performed on all patients who transitioned from the induction trial. Baseline data from the induction trial were used as the reference for change from baseline assessments.
The adjusted proportion of patients with CRR at Year 1 and the proportion of patients with CRR without renal flares at Year 2 were generated using a logistic regression model. Factors in the model included treatment, and the covariates race (Asian vs non-Asian) and proteinuria at screening (UP/UC <3 vs ≥ 3). The same model was used to generate the adjusted proportions of patients with cCRR at Year 1 and cCRR without renal flares at Year 2. Descriptive analyses of AEs were performed for the treated set.
When the primary endpoint of the induction trial was not met, an unblinding of treatment codes for patients continuing in the maintenance trial was performed to allow benefit–risk discussions between investigators and patients. The partial unblinding was not expected to have a meaningful impact on the primary endpoint derived from laboratory measurements, but could have had some impact on patient-reported outcomes (Supplemental Appendix S1).
Results
Patient disposition and baseline characteristics
Of the 121 patients treated in the induction trial, 69 patients (57.0%) completed 1 year of treatment, demonstrated either a CRR, a PRR or proteinuria ≤1.0 g/day (or UP/UC ≤1), consented to continue treatment and were transitioned to the maintenance trial (Figure 1). Patient flow from the 1293.10 induction trial to the 1293.13 maintenance trial *Patients who reached either CRR, PRR or proteinuria ≤1 g/day (or UP/UC ≤1) at Year 1. †Patients continued the treatment they were randomised to in the 1293.10 trial. ‡Four placebo patients discontinued trial medication after partial unblinding, but they continued trial participation and were considered compliant; one patient moved to another state. AE, adverse event; CRR, complete renal response; PRR, partial renal response; UP/UC, urinary protein/creatinine ratio.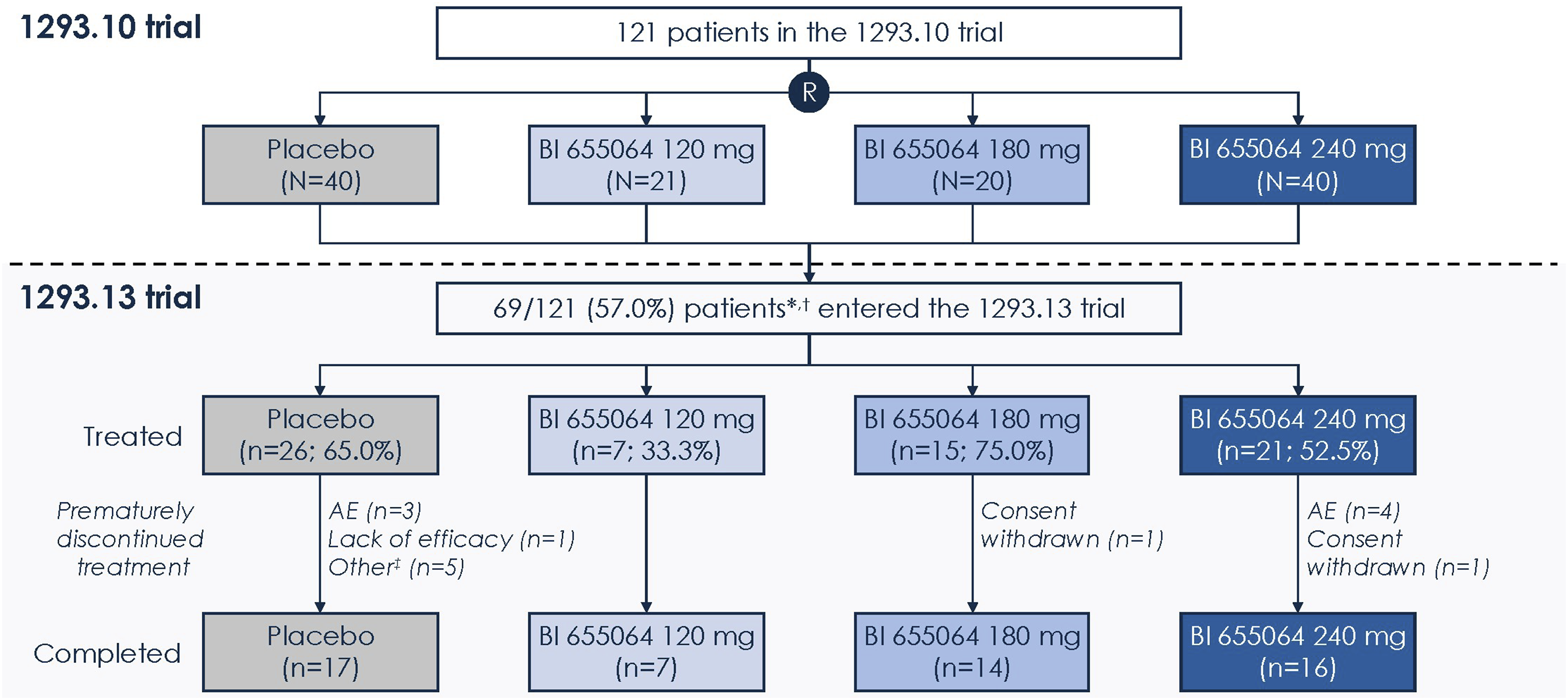
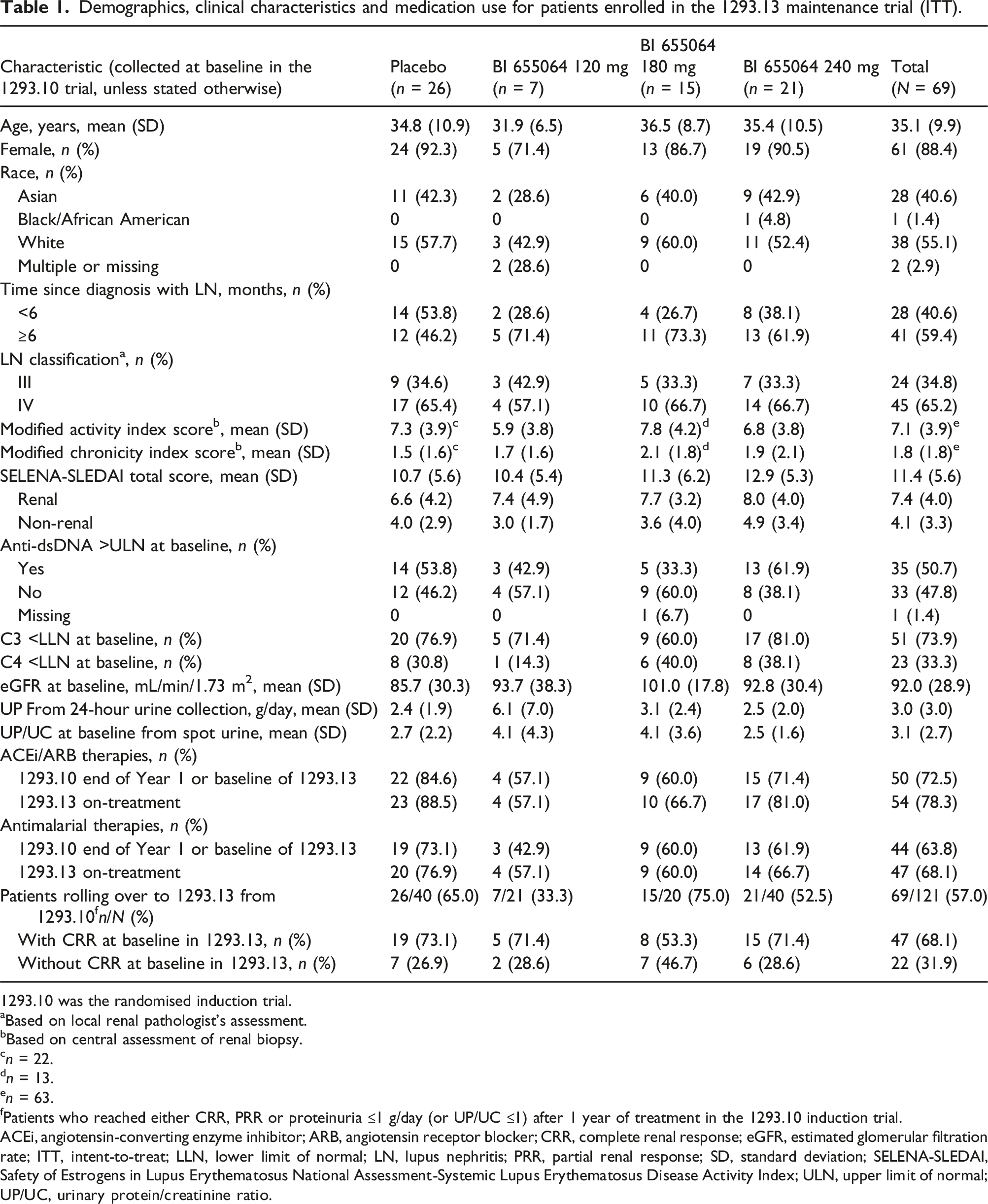
Demographics, clinical characteristics and medication use for patients enrolled in the 1293.13 maintenance trial (ITT).
1293.10 was the randomised induction trial.
aBased on local renal pathologist’s assessment.
bBased on central assessment of renal biopsy.
cn = 22.
dn = 13.
en = 63.
fPatients who reached either CRR, PRR or proteinuria ≤1 g/day (or UP/UC ≤1) after 1 year of treatment in the 1293.10 induction trial.
ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; CRR, complete renal response; eGFR, estimated glomerular filtration rate; ITT, intent-to-treat; LLN, lower limit of normal; LN, lupus nephritis; PRR, partial renal response; SD, standard deviation; SELENA-SLEDAI, Safety of Estrogens in Lupus Erythematosus National Assessment-Systemic Lupus Erythematosus Disease Activity Index; ULN, upper limit of normal; UP/UC, urinary protein/creatinine ratio.
The total population of the induction trial 14 and the subgroup who transitioned to the maintenance trial had similar baseline characteristics. Imbalances in baseline demographics and clinical characteristics from the induction trial 14 carried over to the maintenance trial. More patients receiving placebo had been diagnosed with LN <6 months prior to enrolment (53.8% vs 26.7%–38.1% for those receiving BI 655064). Concomitant ACEi/ARB and antimalarial drugs, which could have favourably impacted renal response, were used more frequently in the BI 655064 240 mg and placebo groups versus the other treatment groups, in both Years 1 and 2 (Table 1). Haematuria was detected at baseline of the 2-year treatment period in a comparable proportion of patients in the BI 655064 groups (40.0%–52.4%) and the placebo group (42.3%) (Table S2).
Efficacy
Patients with CRR
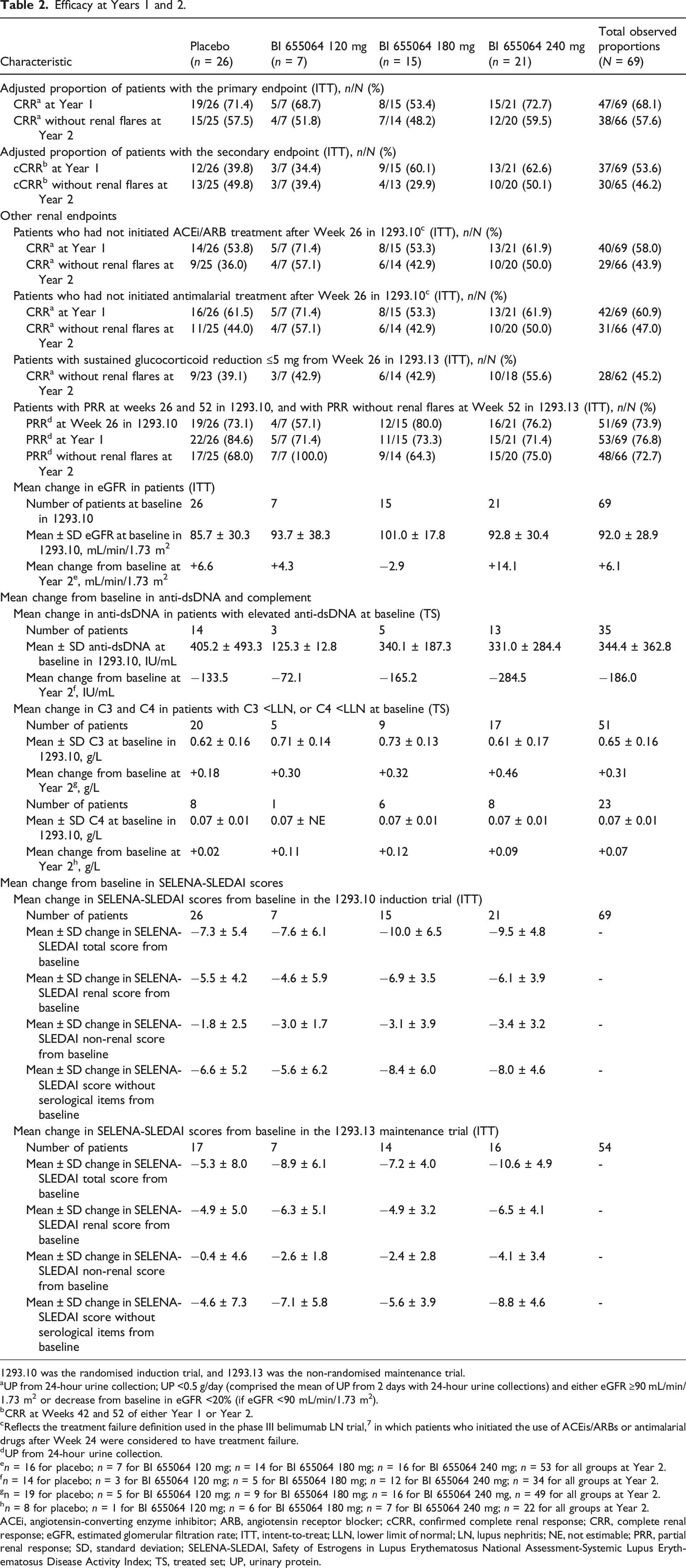
Efficacy at Years 1 and 2.
1293.10 was the randomised induction trial, and 1293.13 was the non-randomised maintenance trial.
aUP from 24-hour urine collection; UP <0.5 g/day (comprised the mean of UP from 2 days with 24-hour urine collections) and either eGFR ≥90 mL/min/1.73 m2 or decrease from baseline in eGFR <20% (if eGFR <90 mL/min/1.73 m2).
bCRR at Weeks 42 and 52 of either Year 1 or Year 2.
cReflects the treatment failure definition used in the phase III belimumab LN trial, 7 in which patients who initiated the use of ACEis/ARBs or antimalarial drugs after Week 24 were considered to have treatment failure.
dUP from 24-hour urine collection.
en = 16 for placebo; n = 7 for BI 655064 120 mg; n = 14 for BI 655064 180 mg; n = 16 for BI 655064 240 mg; n = 53 for all groups at Year 2.
fn = 14 for placebo; n = 3 for BI 655064 120 mg; n = 5 for BI 655064 180 mg; n = 12 for BI 655064 240 mg; n = 34 for all groups at Year 2.
gn = 19 for placebo; n = 5 for BI 655064 120 mg; n = 9 for BI 655064 180 mg; n = 16 for BI 655064 240 mg, n = 49 for all groups at Year 2.
hn = 8 for placebo; n = 1 for BI 655064 120 mg; n = 6 for BI 655064 180 mg; n = 7 for BI 655064 240 mg; n = 22 for all groups at Year 2.
ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; cCRR, confirmed complete renal response; CRR, complete renal response; eGFR, estimated glomerular filtration rate; ITT, intent-to-treat; LLN, lower limit of normal; LN, lupus nephritis; NE, not estimable; PRR, partial renal response; SD, standard deviation; SELENA-SLEDAI, Safety of Estrogens in Lupus Erythematosus National Assessment-Systemic Lupus Erythematosus Disease Activity Index; TS, treated set; UP, urinary protein.
The overall adjusted proportion of patients reaching cCRR at Years 1 and 2 decreased from 53.6% to 46.2% (Table 2). The adjusted proportion of patients reaching cCRR at Years 1 and 2, respectively, decreased from 60.1% to 29.9% in the BI 655064 180 mg group and from 62.6% to 50.1% in the 240 mg group, whereas cCRR was largely unchanged (34.4% to 39.4%) in the 120 mg group and increased from 39.8% to 49.8% in the placebo group. Median changes from baseline for UP/UC (from spot urine) decreased during Year 1, with the greatest decrease in the BI 655064 180 mg group (Figure S2). During Year 2, median changes from baseline for UP/UC (from spot urine) generally stabilised in all treatment groups, with the BI 655064 180 mg group continuing to show the largest median reduction in UP/UC.
Table 2 (other renal endpoints) summarises the data for patients with CRR without renal flares who had not initiated ACEi/ARB or antimalarial treatment after Week 26 in the induction trial; patients with CRR and sustained glucocorticoid reduction ≤5 mg from Week 26 in the maintenance trial; and patients with PRR without renal flares.
At Year 2, there was a considerable increase in eGFR from baseline in the BI 655064 240 mg group (mean change from baseline: +14.1 mL/min/1.73 m2); a slight increase in the BI 655064 120 mg group (+4.3 mL/min/1.73 m2) and the placebo group (+6.6 mL/min/1.73 m2); but no improvement in the BI 655064 180 mg group (−2.9 mL/min/1.73 m2) (Table 2).
In patients who were anti-dsDNA-positive at baseline, the most prominent decrease in mean anti-dsDNA levels at Year 2 was reported in the BI 655064 240 mg and 180 mg groups (−284.5 and −165.2 IU/mL, respectively) versus the placebo and BI 655064 120 mg groups (−133.5 and −72.1 IU/mL, respectively). At Year 2, increases in C3 and C4 levels (in patients with baseline C3/C4 levels below the lower limit of normal) were higher in the BI 655064 treatment groups (+0.30 to +0.46 g/L for C3; +0.09 to +0.12 g/L for C4) versus the placebo group (+0.18 g/L for C3; +0.02 g/L for C4) (Table 2).
Change in SELENA-SLEDAI scores from baseline in the induction trial
Following 2 years of treatment, numerically greater improvements in SELENA-SLEDAI scores (Figure 2 and Table 2) were observed in the BI 655064 groups compared with placebo. At Year 2, the mean decrease from baseline in SELENA-SLEDAI total score was greatest in the BI 655064 240 mg group (−10.6 points), followed by the 120 and 180 mg groups (−8.9 and −7.2 points, respectively) and the placebo group (−5.3 points) (Figure 2(a)). In all treatment groups, SELENA-SLEDAI renal scores decreased from baseline during Year 1 and remained relatively stable during Year 2 (Figure 2(b)). At Year 2, the mean decreases in SELENA-SLEDAI renal scores from baseline were greater in the BI 655064 120 and 240 mg groups (−6.3 and −6.5 points, respectively) than the 180 mg and placebo groups (−4.9 points in each group). Notably, the mean SELENA-SLEDAI renal score at baseline was lowest in the placebo group (6.6 points) compared with all other treatment groups (7.4–8.0) (Table 1). After 2 years of treatment, haematuria was present in 0/7 (0%), 1/14 (7.1%) and 1/16 (6.3%) patients in the BI 655064 120 mg, 180 mg and 240 mg groups, respectively, compared with 0/17 (0%) patients in the placebo group (Table S2). Reductions in the mean SELENA-SLEDAI non-renal scores from baseline in all BI 655064 groups were consistently observed during Year 1 (−3.0 to −3.4 points), with changes in scores stabilising at low levels during Year 2 (−2.4 to −4.1 points) (Figure 2(c)). Conversely, mean SELENA-SLEDAI non-renal scores decreased from baseline in the placebo group during Year 1 (−1.8 points) but increased by 1.4 points during Year 2. Similar trends were observed in the SELENA-SLEDAI score items not requiring serological parameters at Year 2 (Figure 2(d)), with smaller mean decreases from baseline in the BI 655064 180 mg and placebo groups (−5.6 and −4.6 points, respectively) than the BI 655064 120 and 240 mg groups (−7.1 and −8.8 points, respectively). At Year 2, more patients in the BI 655064 treatment groups reached a four-point (76.2%–85.7%), five-point (61.9%–71.4%) and six-point (57.1%–61.9%) improvement in SELENA-SLEDAI total score versus the placebo group (38.5%–42.3%) (Figure 2(e) to (g)). Change in SELENA-SLEDAI scores from baseline in the 1293.10 induction trial at Years 1 and 2 for patients who rolled over to the 1293.13 maintenance trial (ITT). (A) Total score*; (B) renal score†; (C) non-renal score‡; (D) score without serological items§; and the proportion of patients with: (E) four-point, (F) five-point and (G) six-point changes from baseline (ITT) *Mean baseline values in 1293.10 were: 10.7 (placebo), 10.4 (BI 655064 120 mg), 11.3 (BI 655064 180 mg) and 12.9 (BI 655064 240 mg). †Mean baseline values in 1293.10 were: 6.6 (placebo), 7.4 (BI 655064 120 mg), 7.7 (BI 655064 180 mg) and 8.0 (BI 655064 240 mg). ‡Mean baseline values in 1293.10 were: 4.0 (placebo), 3.0 (BI 655064 120 mg), 3.6 (BI 655064 180 mg) and 4.9 (BI 655064 240 mg). §Mean baseline values in 1293.10 were: 8.0 (placebo), 8.4 (BI 655064 120 mg), 9.2 (BI 655064 180 mg) and 10.0 (BI 655064 240 mg).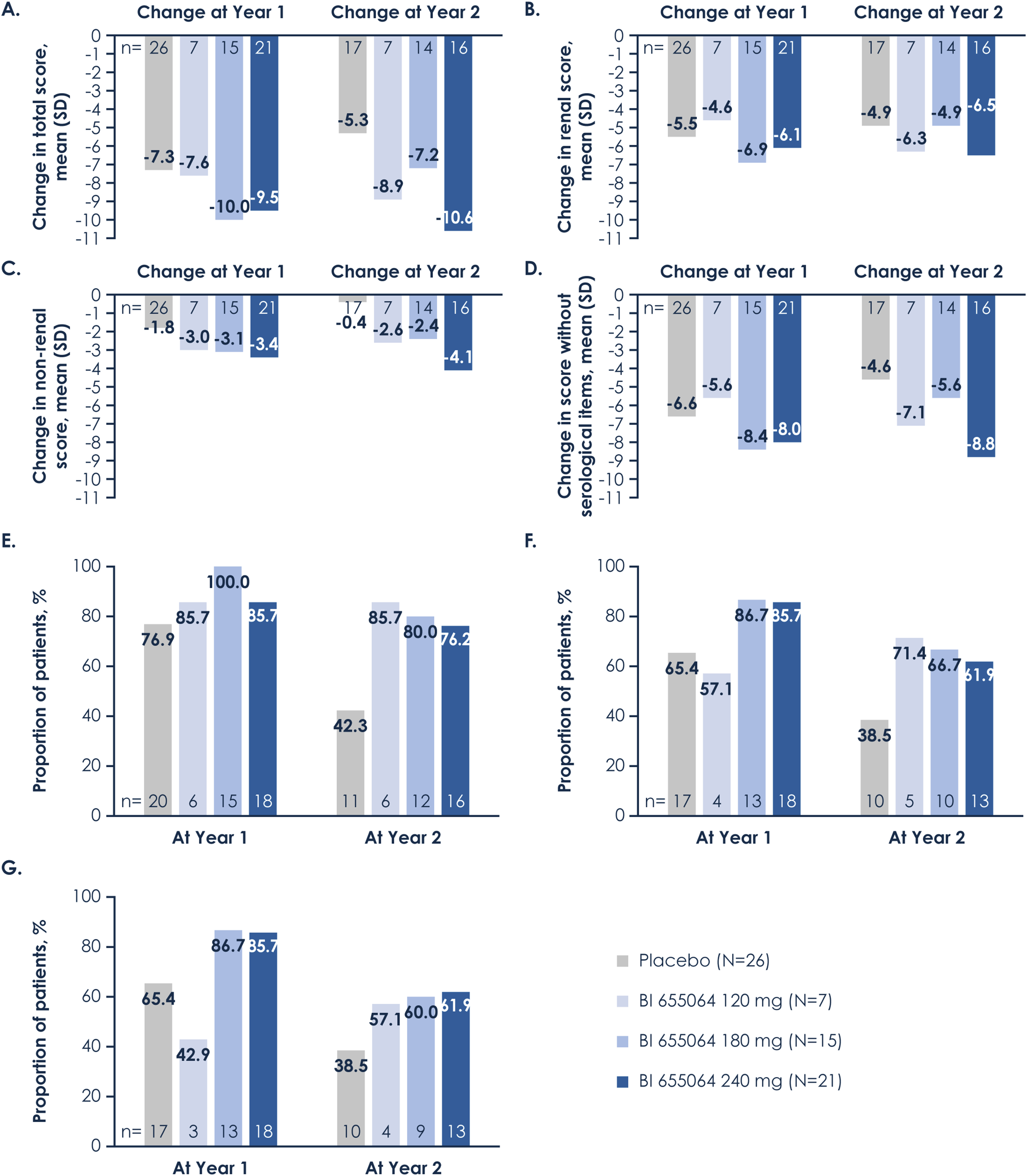
Supplemental Appendix S1 summarises data for LupusPRO and FACIT-F scores (Table S4), showing numerically greater improvement in LupusPRO HRQoL and non-HRQoL total scores among patients in the BI 655064 treatment groups versus the placebo group (Figure S3).
Assessment of BI 655064 plasma concentration after completion of the trial showed unexpectedly low levels of BI 655064, that was <2 µg/mL, or below the limit of quantification, in several patients (Figure 3), which may suggest a lack of adherence to treatment despite patients reporting that they were self-administering treatment. BI 655064 plasma concentrations in the (A) 120 mg, (B) 180 mg and (C) 240 mg groups (pharmacokinetics analysis set*) Dashed line indicates projected steady-state trough concentration of 10 µg/mL at BI 655064 120 mg, 20 µg/mL at BI 655064 180 mg and 30 µg/mL at BI 655064 240 mg. *Included all patients from the treated set who provided ≥1 evaluable observation for a pharmacokinetics endpoint that was not excluded due to a protocol deviation relevant to the evaluation of pharmacokinetics.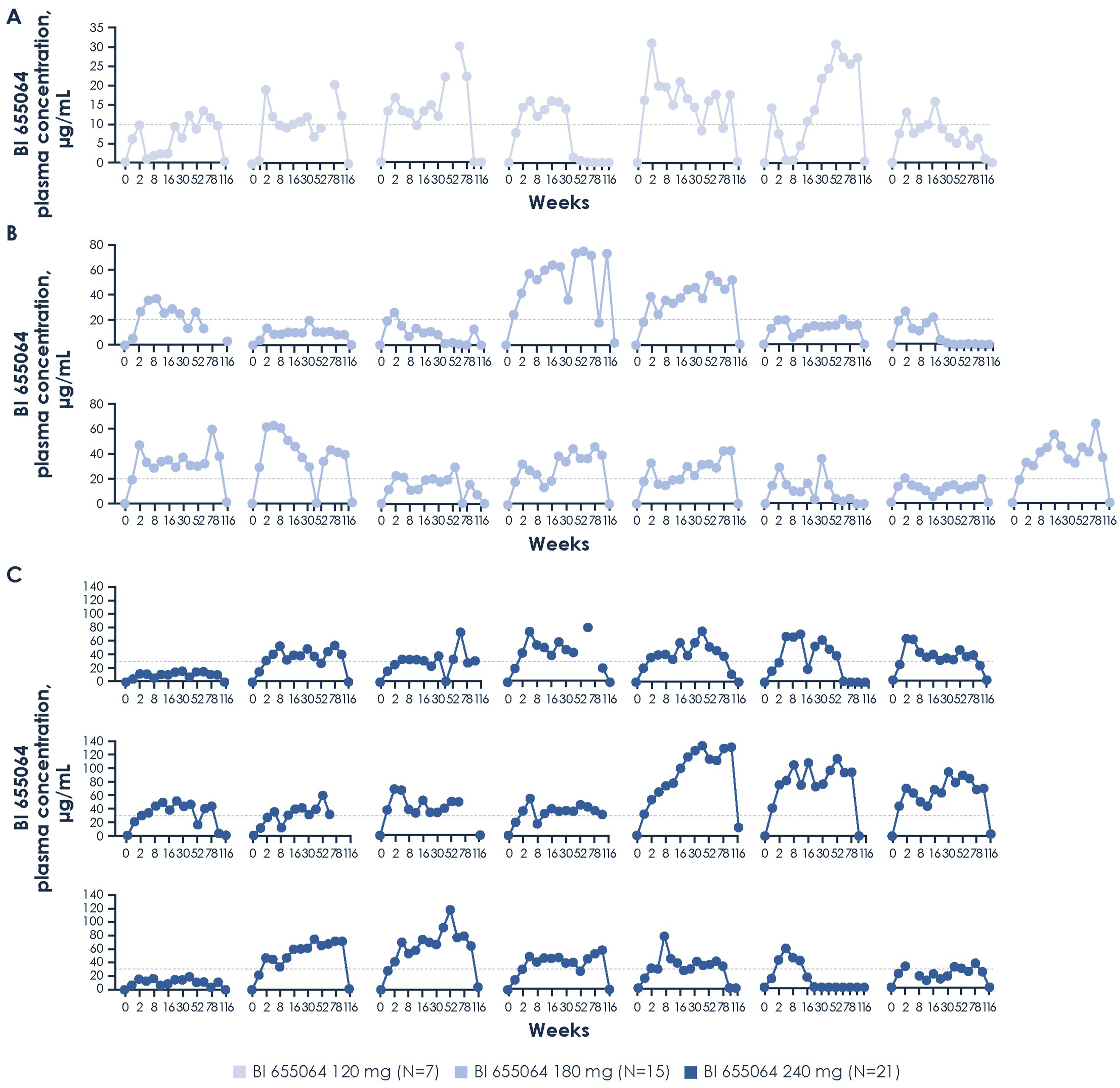
Safety
The safety data need to be considered in the context of the between-group imbalances in the proportions of patients rolling over for a second year of treatment with BI 655064 120 (7/21; 33.3%), 180 (15/20; 75.0%), 240 mg (21/40; 52.5%) or placebo (26/40; 65.0%) in the maintenance trial.
Summary of safety data after 2 years of treatment, and during Years 1 and 2 (TS).
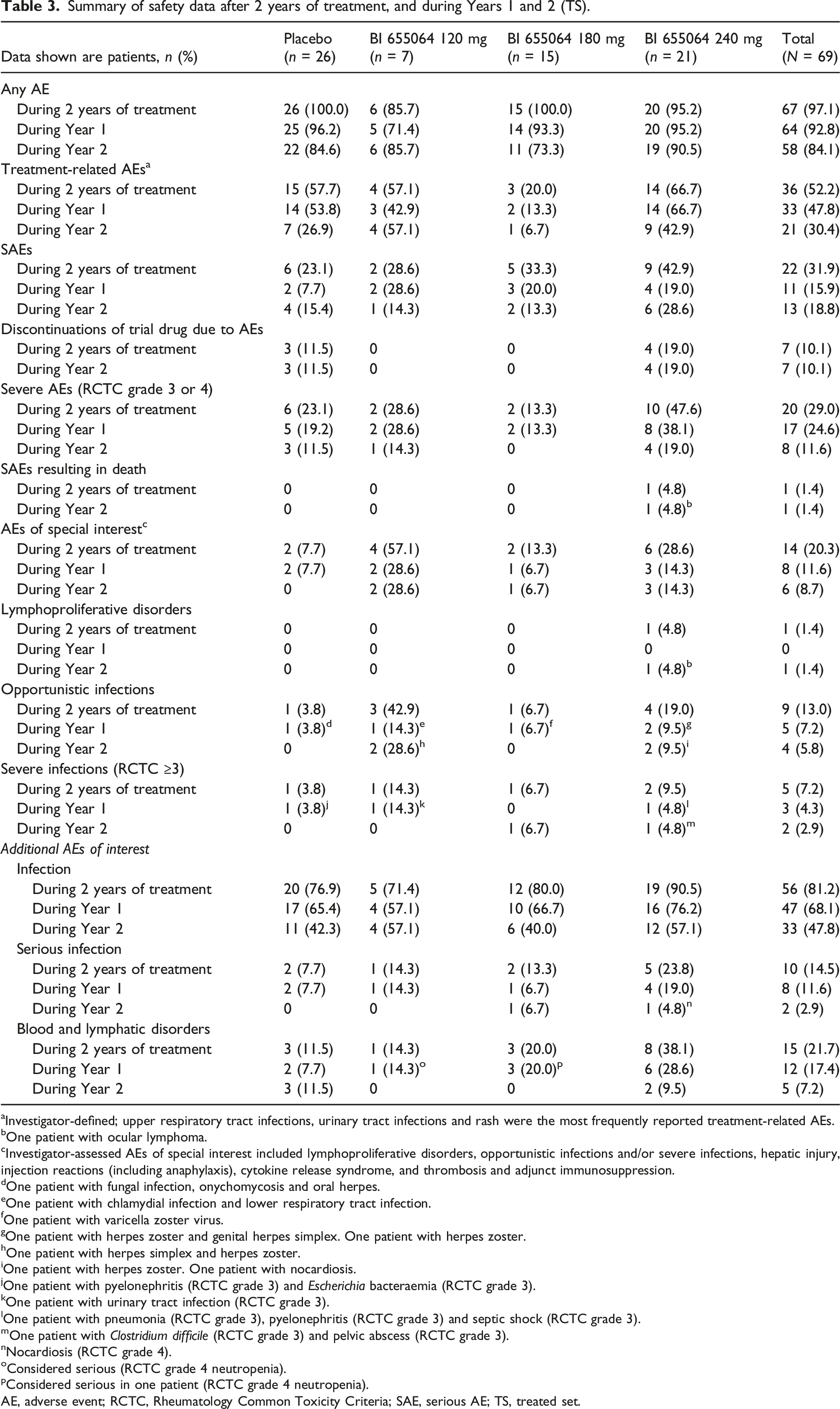
aInvestigator-defined; upper respiratory tract infections, urinary tract infections and rash were the most frequently reported treatment-related AEs.
bOne patient with ocular lymphoma.
cInvestigator-assessed AEs of special interest included lymphoproliferative disorders, opportunistic infections and/or severe infections, hepatic injury, injection reactions (including anaphylaxis), cytokine release syndrome, and thrombosis and adjunct immunosuppression.
dOne patient with fungal infection, onychomycosis and oral herpes.
eOne patient with chlamydial infection and lower respiratory tract infection.
fOne patient with varicella zoster virus.
gOne patient with herpes zoster and genital herpes simplex. One patient with herpes zoster.
hOne patient with herpes simplex and herpes zoster.
iOne patient with herpes zoster. One patient with nocardiosis.
jOne patient with pyelonephritis (RCTC grade 3) and Escherichia bacteraemia (RCTC grade 3).
kOne patient with urinary tract infection (RCTC grade 3).
lOne patient with pneumonia (RCTC grade 3), pyelonephritis (RCTC grade 3) and septic shock (RCTC grade 3).
mOne patient with Clostridium difficile (RCTC grade 3) and pelvic abscess (RCTC grade 3).
nNocardiosis (RCTC grade 4).
oConsidered serious (RCTC grade 4 neutropenia).
pConsidered serious in one patient (RCTC grade 4 neutropenia).
AE, adverse event; RCTC, Rheumatology Common Toxicity Criteria; SAE, serious AE; TS, treated set.
Infections were the most frequently reported AE during 2 years of treatment, experienced by 71.4%, 80.0% and 90.5% of patients in the BI 655064 120, 180 and 240 mg groups, respectively, versus 76.9% in the placebo group (Table 3 and Table S5). Overall, more patients experienced infections during Year 1 (68.1%) than Year 2 (47.8%), except for the BI 655064 120 mg group, which remained unchanged (Table 3). Serious infections occurred in four patients in the BI 655064 240 mg group (19.0%), in one patient in each of the BI 655064 120 and 180 mg groups (14.3% and 6.7%) and in two patients in the placebo group (7.7%) at Year 1, and in one patient each in the BI 655064 180 and 240 mg groups (6.7 and 4.8%) at Year 2. There were three patients with low absolute neutrophil count (ANC; <1000/µL) during Year 1 in the BI 655064 240 mg group compared with 0–1 patient in the other treatment groups; only one patient in the placebo group had low ANC during Year 2. Notably, patients with low ANC did not develop severe infections. One patient in the BI 655064 240 mg group died during Year 2 from ocular lymphoma, which was considered treatment-related according to the investigator (Supplemental Appendix S1). There were no thrombotic events, hepatic events, cytokine release syndrome or injection site reactions.
Discussion
In this exploratory, phase II maintenance trial, BI 655064 failed to demonstrate efficacy benefits versus placebo during a second year of treatment for the primary endpoint (CRR without renal flares at Year 2). However, there was a numerically greater effect of BI 655064 versus placebo in some analyses on renal and non-renal endpoints, particularly in SELENA-SLEDAI total scores and subscores.
In patients with LN, changes in SELENA-SLEDAI total score are mainly driven by the renal components, whereas changes in the non-renal subscores tend to be small. Although this trial was not powered to show a statistically significant difference versus the placebo group, absolute differences in the non-renal subscore for the BI 655064 180 and 240 mg groups were 1.3 and 1.6 at Year 1, and 2.0 and 3.7 at Year 2, respectively. These differences are numerically greater than the changes in non-renal SLEDAI-2K subscores seen with anifrolumab in the phase II TULIP-LN trial, where the maximum difference between anifrolumab and placebo was 1.3 at Year 1 and 1.3 at Year 2.17,18
The presence or absence of haematuria was reported semi-quantitatively as part of the SELENA-SLEDAI renal scores. In this worldwide study, urine analysis was carried out by a central laboratory at each visit, but a more detailed analysis of fresh urine could not be performed due to transportation time. The facilities to test for acanthocytes, which are considered the most characteristic form of dysmorphic red blood cells suggestive of glomerular haematuria, 19 were not established at all local participating sites. As shown by analyses from the Euro-Lupus Nephritis Trial and MAINTAIN Nephritis Trial, the inclusion of haematuria as a renal response criterion in LN did not improve the prognostic value of proteinuria.20,21 Following discussions with key experts, it was agreed not to utilise haematuria as a component of the response criteria in our trials.
Just over half of patients (57%) entered the maintenance trial from the induction trial. This drop-out rate could be attributed to the fact that patients were required to meet the inclusion criteria of having completed the induction trial, responded to treatment at Year 1, and consented to continue treatment in the maintenance trial. In addition, a potential confounder in this maintenance trial was that it included a population with differently defined responses at entry, as patients rolling over could have had CRR, PRR or proteinuria ≤1.0 g/day (or UP/UC ≤1) at the end of the induction trial. More patients rolling over in the 180 mg group had not reached CRR at baseline (46.7% vs 26.9%–28.6% in the other groups). Future studies should consider more homogeneous response criteria between trials.
Reasons as to why the induction trial did not meet its primary endpoint have been discussed previously, 14 including compliance, confounders, and the high placebo response rate. Pharmacokinetic analysis performed in this trial suggested a lack of adherence to BI 655064 treatment, despite patients reporting that they were self-administering treatment. Suspected non-adherence in both the induction and the maintenance trial may have further impacted BI 655064 exposure in the maintenance trial. As the induction and maintenance trials were both double-blind, the potential non-adherence was only suspected post study completion. Weekly self-administered injections may have been perceived as a considerable treatment burden by patients, illustrated by the high drop-out rate between the induction and maintenance trials. In an ideal situation, injections would always be carried out under supervision, but it would have been unrealistic to demand that patients visit the participating sites on a weekly basis. It might have improved compliance if, on the days of the regular visits, patients carried out the injections on site. Low MMF levels were also observed in some of the patients with suspected non-adherence. As MMF is part of the SoC, regular evaluation of MMF levels could be used as a surrogate measure of compliance in future trials. In addition, 24-hour urine collections were incomplete in some cases, with recorded volumes being conspicuously small. To account for this, CRR was analysed using UP/UC from spot urine collections.
Similarly, in the maintenance trial, the use of ACEis/ARBs and antimalarial drugs were potential confounders. More patients in the placebo group versus the BI 655064 groups were treated with ACEis/ARBs (84.6%–88.5% vs 57.1%–81.0%) or antimalarial drugs (73.1%–76.9% vs 42.9%–66.7%). When excluding patients who started ACEis/ARBs or antimalarial treatment from Week 26 in the induction trial, the proportion of patients with CRR without renal flares decreased from Year 1 to Year 2: from 53.8% to 36.0% (using ACEi/ARB as an exclusion criterion) and 61.5% to 44.0% (using antimalarials as an exclusion criterion) in the placebo group, and from 53.3%–71.4% to 42.9%–57.1% (using both ACEi/ARB and antimalarials as exclusion criteria) in the BI 655064 groups. Thus, baseline ACEi/ARB and antimalarial use may have increased responses in the placebo group during Year 2. A higher-than-expected response rate in the placebo group may also be due to these patients being more likely to respond to SoC and/or having more reversible disease, with more than half of the placebo group diagnosed with LN for <6 months (53.8%), versus 26.7%–38.1% in the BI 655064 groups. Patients receiving placebo also had a numerically lower mean modified chronicity index score and mean SELENA-SLEDAI renal subscore at baseline than those receiving BI 655064 (1.5 vs 1.7–2.1 and 6.6 vs 7.4–8.0, respectively). Other potential causes of the unexpected high response rate in the placebo group have been discussed previously. 14
This exploratory maintenance trial provided insights into the long-term safety profile of CD40 blockade as add-on to SoC. Safety data over 2 years confirmed the safety and tolerability profile of BI 665064 in the total population of the induction trial following 1 year of treatment, 14 with fewer AEs observed during Year 2 than Year 1. During Year 1, a potential risk for serious infections with BI 655064 240 mg was noted 14 ; however, during Year 2, this imbalance across treatment groups was not observed. Caution must be exercised in drawing definitive conclusions due to the differing proportions of patients per treatment group compared with the induction trial, as well as the overall small patient numbers. An independent Data Monitoring Committee oversaw safety throughout the induction and maintenance trials and there was no recommendation to discontinue treatment.
Other second-generation anti-CD40/CD40L antibodies are also in development. Recently, the anti-CD40L antagonist dapirolizumab pegol demonstrated significant improvement in disease activity in a phase III trial involving patients with moderate-to-severe SLE. 22 Phase II trials of iscalimab (CFZ533) in patients with SLE and LN are currently ongoing.23,24
In conclusion, although this exploratory, phase II maintenance trial failed to demonstrate the benefits of BI 655064 in renal outcomes, there were numerical improvements observed in non-renal aspects of SLE versus placebo. Clinical trials in SLE and LN are challenging, and further work is needed to address these challenges.
Supplemental Material
Supplemental Material - Two-year treatment experience with BI 655064, an antagonistic anti-CD40 antibody, in patients with active lupus nephritis: An exploratory, phase II maintenance trial
Supplemental Material for Two-year treatment experience with BI 655064, an antagonistic anti-CD40 antibody, in patients with active lupus nephritis: An exploratory, phase II maintenance trial by Richard Furie, Jürgen Steffgen, Nora Fagan, Juanita Romero-Diaz, Yingyos Avihingsanon, Dimitrios T Boumpas, Kajohnsak Noppakun, Jing Wu, Ivette Revollo and David R Jayne in Lupus
Footnotes
Acknowledgments
The authors are grateful to the patients and investigators who participated in this trial. Carolyn Bowler, PhD, CMPP, of Hyperion, OPEN Health Communications (London, UK), provided medical writing, editorial and formatting support; Yuheng Ouyang, of Nucleus Global provided editorial support, both of which were contracted and funded by Boehringer Ingelheim. The authors also thank Ramtej Dasari of Boehringer Ingelheim, US, who provided project programming support, and Simone Deutschel of Boehringer Ingelheim, RCV Austria, who provided operational support of the trial.
Declaration of conflicting interests
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors did not receive payment related to the development of this manuscript. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy, as well as intellectual property considerations. The study was supported and funded by Boehringer Ingelheim. RF and YA have served as consultants for Boehringer Ingelheim. JS, NF, JW and IR are employees of Boehringer Ingelheim. JS is listed as inventor on certain patent applications related to BI 655064. JR-D has received consulting fees and support for attending meetings from Boehringer Ingelheim. DTB reports no conflicts of interest. KN has received grants from AstraZeneca, Boehringer Ingelheim, Novo Nordisk, Omeros and Visterra Inc.; has received consulting fees from AstraZeneca, Bayer, Novo Nordisk and Takeda; has received honoraria from Abbott, Astellas Pharma, AstraZeneca, Bayer, Boehringer Ingelheim, Celltrion, Kyowa Kirin, Novartis, Organon, Roche, Sanofi-Aventis, Siam Pharmaceuticals, Takeda, Thai Otsuka, Viatris and Zuellig Pharma; and is a board member of the Thai Transplant Society. DRJ was supported by the National Institute for Health and Care Research Cambridge Biomedical Research Centre, has received consulting fees from AstraZeneca, Boehringer Ingelheim, ChemoCentryx, GSK, Novartis, Takeda and Vifor, and owns stock or stock options in Aurinia Pharmaceuticals.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Boehringer Ingelheim.
Contributorship
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published.
Trial registration number
NCT03385564.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.