
Abstract
We describe a female patient with systemic lupus erythematosus (SLE) also diagnosed with Fabry’s disease and anti-phospholipid antibody syndrome (APS). SLE and Fabry’s disease are both systemic diseases with variable clinical presentations. Recent studies have shown a relatively high incidence of late onset Fabry’s disease in female heterozygous individuals, suggesting that this condition could be under-diagnosed. We discuss a possible association between SLE and Fabry’s disease and consider the role of lipid abnormalities in the pathogenesis of SLE.
Keywords
Introduction
This case describes a female patient with systemic lupus erythematosus (SLE) and antiphospholipid antibody syndrome (APS). The patient went on to develop atypical symptoms of heart disease and was diagnosed with the lysosomal storage disease, Fabry’s disease (FD), which is characterized by the accumulation of lipids in organs and tissues. SLE and FD are both systemic diseases with variable clinical presentations. Recent studies have shown a relatively high incidence of late onset FD in female heterozygous individuals, suggesting that this condition could be under-diagnosed. Interestingly, the pattern of organ involvement in patients with SLE and FD can be similar and this case highlights similarities in disease pathogenesis between the two conditions. We discuss a possible association between SLE and FD and consider the role of lipid abnormalities in the pathogenesis of SLE.
Case presentation
A 38-year-old Caucasian woman presented with exertional chest pain with shortness of breath.
History of present illness
The patient was diagnosed with SLE in 1987 with an unusual presentation including arthralgia and loose stools. At diagnosis her autoimmune profile was diffuse pattern antinuclear antibody 1/640, positive anti-double-stranded (ds)DNA, negative extractable nuclear antigens (ENA, including anti-Ro) and low complement C3 and C4. Antiphospholipid antibodies (aPL) were negative at diagnosis but became positive in 1996. She had a history of photosensitivity, malar rash, occasional mouth ulcers, increased sweating and joint pain (knees, hands and spine). She had no constitutional symptoms or history suggestive of active infection and no neurological symptoms, cataract, tinnitus, hearing loss or angiokeratoma. Her father died at age 65 with congestive cardiac failure and her mother was alive at age 67 having had a subarachnoid haemorrhage but she had no other cardiovascular risk factors (non-smoker with normal serum cholesterol). The patient had a history of secondary Sjögren’s syndrome, sensory epilepsy and migraine.
Treatment included hydroxycholoroquine (1987–2007) 400 mg/day, azathioprine (from 1990) 100 mg and prednisolone 5 mg. She was also on sodium valproate, cyclizine, amitriptyline, losartan and ibuprofen and had been reviewed routinely by a cardiologist in 1988 with no evidence of heart disease and a normal electrocardiogram (ECG). In a second review in 1999 the echocardiogram demonstrated normal posterior and inter-ventricular septum wall thickness of 0.9 cms. The valves were normal and the function was good at that stage.
In April 2002 she presented with shortness of breath and exertional chest pain when walking upstairs or on inclines. The pain was central and radiated to the left arm. She also complained of palpitations, describing a ‘pounding sensation’ which improved with losartan. She gave no history of syncope but occasionally complained of presyncope when standing up rapidly, and tiredness. In June 2002 she had distinct changes in her exercise capacity and was moderately hypertensive; she was referred to a cardiologist.
Physical examination
She was afebrile and moderately hypertensive (140/70), pulse was 90, regular and venous pressure was elevated. Other vital signs were stable. She weighed 80 kg and her height was 167.2 cm. There was peripheral oedema but no clubbing. She had a butterfly facial rash but no angiokeratoma. Joint exam was normal with no evidence of synovitis. The reflexes were normal and symmetrical and there was no sensory deficit. There was 2/6 ejection systolic murmur at the left sternal edge and basal crepitations on chest examination. There was no hepatosplenomegaly.
Investigations
Her routine full blood count, urea, electrolytes, liver functions, thyroid function and urinary protein creatinine ratio were normal. ECG showed marked QRS changes with hypertrophy, mild QRS broadening and widespread anterolateral ST segment abnormalities. There was sinus rhythm with voltage criteria for left ventricle hypertrophy and normal PR interval. Echocardiogram demonstrated a small left ventricular (LV) cavity with concentric LV hypertrophy with a maximal thickness of 1.7 cm. She had complete LV obliteration on apical view but there was no significant outflow gradient. She had evidence of impaired relaxation on the transmitral Doppler. There was also evidence of moderate right ventricular hypertrophy. The valves were thin, mobile with no lesions and no pericardial effusion. Coronary angiogram demonstrated normal coronary arteries.
Diagnosis
Echocardiograph showed cardiomyopathy (CM) changes (concentric hypertrophy with posterior wall and interventricular septum thickness of 2 cm which was absent in 1999 with the thickness being 0.9 cm). The heart biopsy showed no evidence of amyloid. The sarcoplasmic vacuolation and myofibrillary loss of myocytes seen in the heart biopsy was due to glycosphingolipid deposition (Figure 1(a)) and massive deposition of electron-dense glycosphingolipid in the form of myelin figures in the myocyte sarcoplasm (Figure 1(b)). A similar appearance can be observed in hydroxychloroquine-associated CM (hydroxycloroquine inhibits the function of lysosomal enzymes 1 ). However, when myelin figures are present in large numbers, FD should be considered and confirmed by more specific investigations. Heart biopsy from the systemic lupus erythematosus (SLE) patient showing pathology consistent with Fabry’s disease. (a) Haematoxylin and eosin-stained light micrograph section showing sarcoplasmic vacuolation and myofibrillary loss of myocytes. Magnification × 200 and (b) Electron micrograph showing electron-dense glycosphingolipid in the form of myelin figures in the myocyte sarcoplasm.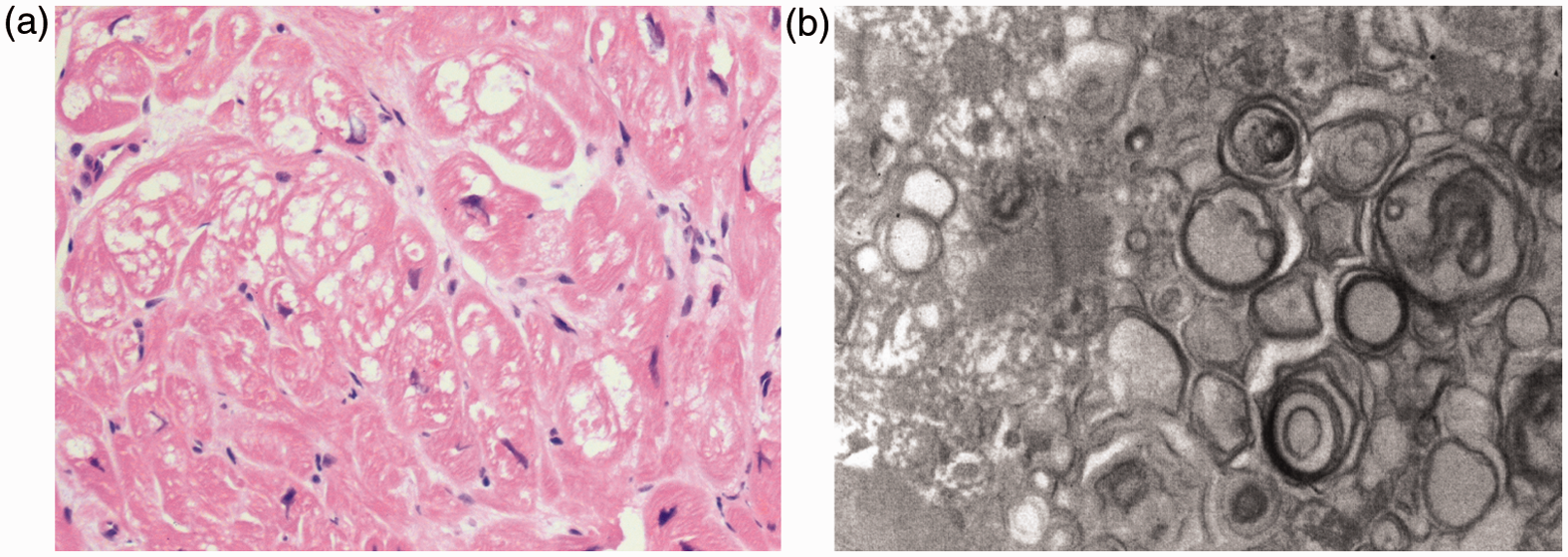
FD is an X-linked deficiency of the enzyme α-galactosidase A (GLA), resulting in accumulation of the glycosphingolipid globotriaosylceramide (Gb3) in organs and tissues. GLA activity was low in this patient at 39 nmol/hr/mg protein (normal range 33–134), and genetic testing in August 2003 showed a rare P343L mutation in the GLA enzyme, confirming the diagnosis.
Treatment and subsequent course
The patient was treated with enzyme-replacement therapy (ERT), agalsidase beta (Fabrazyme), aspirin and diltiazem. Hydroxychloroquine was stopped in November 2007 when a maculopathy was suspected and she has been reviewed regularly since by the ophthalmologists.
In 2008 her echocardiogram showed mild LV dilatation with hypokinesis of the anterior and inferior septum (ejection fraction 50%, LV end diastolic diameter (LVEDD) 53 mm, LV end-systolic diameters (LVESD) 39 mm, left atrium 34 mm, septum 8 mm). Tissue Doppler velocities were reduced at the mitral valve annulus and transmitral inflow confirmed mild diastolic dysfunction. Right heart size and function was normal and there was no relevant valvular abnormality. There was a thin organized layer of pericardial fibrin with a small chronic pericardial effusion. The cardiac magnetic resonance imaging (MRI) showed normal myocardial wall thickness with an ejection fraction of 35%. There was extensive late gadolinium enhancement affecting 50% of the myocardium. It was mainly mid-myocardial and in some areas transmural. There was a small pericardial effusion.
She suffered a myocardial infarction (MI) with a distal anterior descending artery thrombus in June 2009. Her blood tests showed increased IgM aPL 32 mplu/ml (normal range (nr) 0–9) and raised IgM anti-β2 glycoprotein I (β2GPI) antibody 111 U/ml (nr = 0–12). She was treated with atorvastatin (80 mg/day), aspirin and clopidogrel, which was then changed to warfarin (international normalized ratio (INR) 3–4) following discussions with haematologists and rheumatologists. An unstable INR on warfarin treatment, probably linked to variations in her ibuprofen and fluconazole, led to the replacement of warfarin with dalteparin 15,000 units subcutaneously (sc)/day.
In September 2011 she suffered a cerebrovascular accident affecting the right middle cerebral artery whilst on dalteparin. She was started on clopidogrel temporarily and then the dalteparin dose was gradually increased to 8500 units twice daily. Her lupus disease activity remains relatively inactive clinically although aPL, anti-β2 GPI antibodies and lupus anticoagulant continue to be positive. Her dsDNA antibody remained positive (between 160 and 200 IU/ml, normal range ≤50) and complement C3 level varied between 0.80 and 0.96 g/l (normal range 0.9–1.80). Her liver and kidney function have remained stable throughout the course.
Discussion
Differential diagnosis
The clinical presentation could have been due to SLE-related heart disease, APS or atherosclerosis and coronary artery disease (CAD). Rarer causes of heart failure include infection, infiltrative lesions, arrhythmias, drugs and toxins. The coronary angiogram was normal and there was no evidence of hypokinesia or akinesia on echocardiogram. However, the echocardiogram did reveal increased ventricle thickness suggestive of an infiltrative process such as amyloidosis or a storage disease
SLE-related heart disease
Cardiac involvement in lupus is one of the major causes of morbidity and mortality with SLE. It can involve all three layers of the heart – pericardium, myocardium and endocardium. Clinical (symptomatic) pericarditis is seen in approximately 25% of SLE patients during the course of their disease; however, asymptomatic pericardial effusion is more common and is usually detected on echocardiogram. It occurs more frequently at SLE onset or during relapse, although it can occur at any time of the disease. 2 Episodes can be isolated or recurrent 3 and complications include cardiac tamponade, constrictive pericarditis and purulent pericarditis.
Myocarditis is uncommon in SLE, occurring in <10% of patients although subclinical disease is more frequent. 4 Clinical features include dyspnoea, tachycardia, arrhythmias which can progress to ventricular dysfunction, dilated CM and heart failure. There are no typical findings on ECG, and cardiac enzymes may be normal. Large echocardiogram series have found frequencies of global hypokinesis between 5% and 20%. However, segmental areas of hypokinesis can also be indicative of the disease. 5 Magnetic resonance has proved useful in diagnosing myocardial involvement, even at the preclinical stage. 6 However, endomyocardial biopsy remains the investigation of choice.
The notion that lupus myocarditis is an immune complex-mediated disease is supported by immunofluorescence studies demonstrating fine granular immune complex and complement deposition in the walls and perivascular tissues of myocardial blood vessels. Some reports demonstrate an association between anti-SSA/Ro antibodies and myocarditis. 7
Atherosclerosis and CAD
The prevalence of CAD ranges from 6% to 10% in SLE patients and the risk of developing any CAD is four to eight times higher than in control subjects. 8 –11 In young women with SLE, the risk of MI is increased up to 50-fold. 12 Traditional risk factors (hypertension, hyperlipidaemia, smoking, obesity, diabetes mellitus), non-traditional risk factors (e.g. autoimmune inflammation) and corticosteroids increase the risk of CAD.
Heart involvement in APS
Heart valve lesions (vegetation, valve thickening and dysfunction) are frequently reported in patients with APS with and without SLE 13 and in those with aPL alone. 14 Libman-Sacks endocarditis is a feature of APS. SLE and APS have similar valve abnormalities, varying from minimal thickening and/or vegetations to severe valve distortion and dysfunction. Hojnik et al. 14 reviewed echocardiographic studies of primary APS patients and reported 32%–38% prevalence of valve lesions most frequently involving left-sided valves (mitral more common than aortic), whereas Libman-Sacks commonly involves the tricuspid valve. Turiel et al. 15 demonstrated valve abnormalities in 82% of primary APS patients and mitral valve thickening in 63%, using trans-oesophageal echocardiography (TEE). Erdogan et al. 16 demonstrated cardiac involvement in 84% of primary APS patients and mitral regurgitation in 77.4%. Valvular disease is mostly mild and asymptomatic; only rarely (4%–6%) patients develop valve disease severe enough to require surgical treatment.
Atherosclerosis and CAD in APS
APS patients suffer from an increased rate of cardiovascular accidents: It is the presenting manifestation in 2.8% of patients and up to 5.5% of APS patients suffer an MI during the course of their disease. 13 Correlation between serum levels of aPL and anti-β2GPI antibodies and the incidence and severity of acute coronary syndrome, MI and stroke was shown by Veres and others. 17 Lupus anticoagulant and anti-β2GPI antibodies are the risk factors for MI in the assessment of SLE patient cohorts. 18,19
Antimalarial (hydroxychloroquine/chloroquine)-induced CM
According to case reports, clinical heart failure and restrictive CM are extremely rare but potentially life-threatening complications associated with long-term treatment with chloroquine or hydroxychloroquine. 20 –22 Other cardiac complications include myocardial thickening, cardiac insufficiency and conduction disorders. 23 The interval between treatment commencement and development of cardiac complications ranges from seven months to 25 years and the total cumulative doses ranges from 292 g to 4380 g for chloroquine. 24,25
A finding of restrictive physiology on two-dimensional echocardiogram should prompt the clinician to consider various aetiologies and investigate accordingly. The pathologic findings associated with antimalarial cardiotoxicity are similar to FD and secondary amyloidosis. 24,26 –28 Cardiac biopsy with findings such as vacuolar myopathy, myeloid bodies and curvilinear bodies can prove helpful in cases with new onset heart failure. Clinical resolution and significant regression of diastolic dysfunction are demonstrated with discontinuation of chloroquine.
FD
FD is an X-linked deficiency of the lysosomal enzyme GLA resulting in the accumulation of the glycosphingolipid Gb3 in organs and tissues. Accumulation of Gb3 leads to progressive organ failure and premature death although the precise pathogenic mechanisms leading to clinical symptoms are not completely understood. 29 Typical manifestations include angiokeratoma, sweating disturbances, neuropathic pain, hearing impairment as well as heart, kidney and central nervous system involvement. 30 Clinical onset in homozygous males (with very low levels of GLA activity) is severe occurring in childhood; however, heterozygous females (with intermediate GLA enzyme activity) have late onset disease or may be asymptomatic. Incidence of classic onset disease is rare (one in 37,000); however, recent neonatal screening studies show that late onset incidence is higher than expected (one in 3100 live births), indicating that the true prevalence of FD is underestimated. 31 Furthermore, it has been suggested that patients with renal, cerebral and cardiac insufficiency of unknown cause might have FD. 32 Interestingly, the pattern of organ involvement in patients with SLE and FD can be similar. 33
The coexistence of FD and autoimmunity including SLE and rheumatoid arthritis has been described previously. 33 –36 Patients have presented with different symptoms (including eye, renal and cardiac involvement) consistent with the heterogeneous nature of FD. Here we describe a patient with SLE and FD complicated by APS. The patient’s severe myocardial dysfunction, not being consistent with APS or other conditions, led to the diagnosis which was confirmed by testing GLA enzyme activity (which was low, although this is not always the case in patients) and by genetic testing that revealed an unusual GLA polymorphism (P343L) contributing GLA enzyme deficiency. 32
Recently, the patient suffered an MI with a distal anterior descending artery thrombus. Stroke, thrombophilia and the presence of autoantibodies, including ‘lupus-associated’ antibodies to dsDNA, ENA and phospholipids, 37 can be features of patients with FD. Since the prevalence of aPL in Fabry’s patients is up to 45% 37 compared to around 30% in lupus patients, 38 it is difficult to assess whether the presence of aPL in this patient resulted from the FD or the APS. However, it seems likely that high levels of aPL that developed between 2002 and 2009 may well have contributed to the MI suffered in 2009.
Originally it was believed that pathology in FD was caused by increased lipid (Gb3) deposition in various organs. However, recent work suggests that Gb3 is immunogenic and provides a continuous stimulus, potentially by changing the nature of lymphocyte cell membranes, thereby creating an environment able to sustain autoimmune responses. 30 These observations are consistent with what is known about defects in immune cell function in patients with SLE. 39 SLE patients are characterized by an altered composition of lymphocyte cell membrane lipids, including Gb3, that can increase their activation. 39 Furthermore, increased accumulation of glycolipids in the cell membrane of lymphocytes increases oxidative stress and the formation of reactive oxygen species, both factors that influence the inflammatory response and contribute to increased cardiovascular risk. 32 Since patients with SLE are associated with an unexplained increased cardiovascular risk (up to 10 times overall and up to 50 times in female patients aged between 35 to 44, 12,40 there may be a case for screening certain SLE patients for Gb3 and GLA enzyme activity, for example, those with unexplained cardiac symptoms.
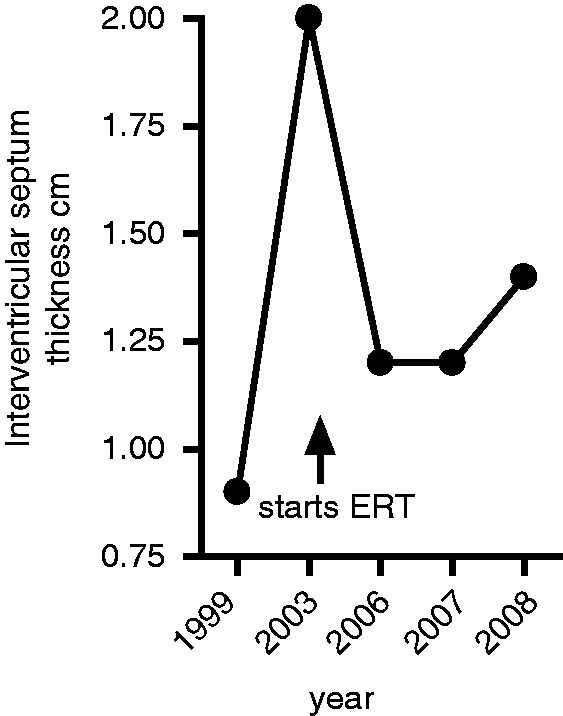
Unlike the previous reports, the patient described here was treated with agalsidase beta, a relatively new therapy for lysosomal storage diseases. 32 Enzyme-replacement therapy (ERT) (agalsidase alfa or agalsidase beta) is able to reduce Gb3 expression in the serum; this was the case in this patient as shown in Figure 2 but its effect on disease symptoms can vary. Both drugs have been shown to improve cardiac involvement in FD, 41,42 which was also the case in our patient (Figure 3). However, as mentioned our patient did go on to have an MI with a distal anterior descending artery thrombus. Relevant to this, one in vitro study reported that ERT may contribute to endothelial damage, which could explain why strokes continue to occur in patients treated with ERT. 43 Reduced serum expression of glycosphingolipid globotriaosylceramide (Gb3) following enzyme-replacement therapy (ERT). Glycosphingolipids (GSLs) were isolated from serum samples taken before and after commencement of ERT and analysed by high-performance liquid chromatography (HPLC). 44 Results were compared to specific GSL standards and the ratio of Gb3 to GM3 (a GSL whose levels are not affected by AGL deficiency) was calculated. (a) HPLC plots from before ERT compared to the GSL standards and (b) plot of the ratio between of Gb3 and GM3 expression in the serum before and after ERT. Interventricular septum thickness is reduced after enzyme-replacement therapy (ERT). Plot of septum thickness levels before and after commencement of ERT.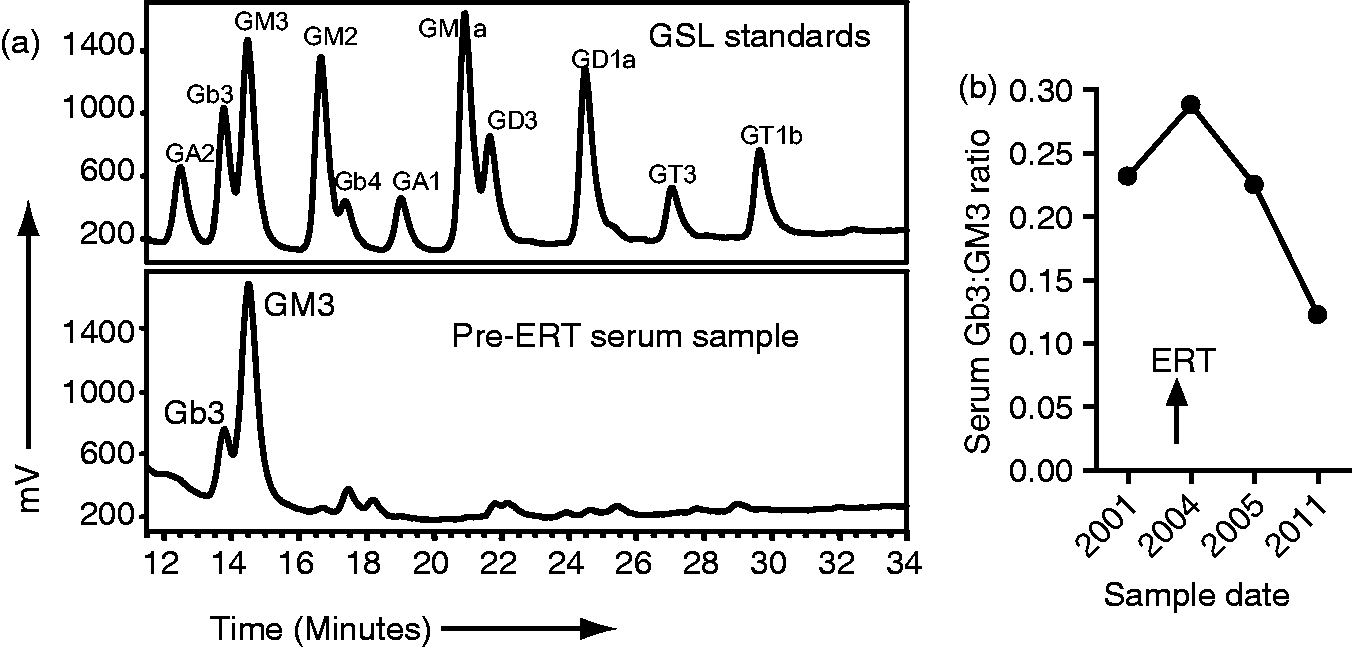
Conclusion
This case highlights similarities in the disease pathogenesis between patients with SLE and FD. The co-existence of FD and SLE may be more common than previously believed. Furthermore, defects in lipid biosynthesis, such as is seen in FD, could contribute to the development of autoimmunity. Finally, this case reminds the physician that although lupus is a complicated disease on its own, approximately 30% of patients also present with a second (or more) concomitant autoimmune disease which adds to the complication. Thus the diagnosis of lupus is just the beginning of the story.
Final diagnosis
SLE and APS with Fabry’s disease.
Ethical approval
The patient provided informed written consent for this case to be presented. Other experimental work was approved by the ethics committee of University College Hospital London (00/0241).
Footnotes
Funding
ECJ is supported by a Arthritis Research UK Career Development Grant (grant number 18106).
Conflict of interest
None declared.