
Abstract
Background
Robinin possesses certain antioxidative and anti-inflammatory properties. However, its mechanisms of action in cardiac hypertrophy (CH) and fibrosis remain inadequately explored.
Methods
An in vitro model of CH was established using angiotensin II-treated rat H9c2 cardiomyocytes treated with escalating concentrations of Robinin. For the in vivo model, pulmonary heart disease-induced CH in rats was induced by chronic hypercapnia and TAC. Myocardial cell markers were evaluated through qRT-PCR, Western blotting, ELISA, cellular immunofluorescence, and HE staining. The role of SIRT1 in Robinin’s effects was determined using pharmacological inhibition with SIRT1-IN-1.
Results
In vitro, Robinin suppressed hypertrophic (ANP, BNP, β-MHC) and fibrotic markers (MyHC, Collagen I, TGF-β1) and reduced NLRP3 components (IL-1β, IL-18, ASC, Caspase-1) and oxidative stress (OS, ↓MDA, ↑SOD/HO-1/Nrf2) in a dose-dependent manner. These effects were mediated by SIRT1 upregulation and NF-κB inactivation. Crucially, SIRT1 inhibition abolished Robinin’s protection against hypertrophy and inflammation. In vivo, Robinin attenuated pulmonary heart disease-induced hypertrophy, fibrosis, NLRP3 activation, and OS while restoring SIRT1/NF-κB balance.
Conclusion
Robinin alleviates CH in pulmonary heart disease by modulating the SIRT1/NF-κB pathway to suppress OS and NLRP3 inflammasome activation, establishing its novel therapeutic potential.
Introduction
Chronic pulmonary heart disease is a prevalent cardiovascular condition in China, characterized by a high mortality rate. 1 It primarily arises due to chronic diseases of the lungs or thorax or lesions in the pulmonary vasculature, leading to pulmonary hypertension and cardiac hypertrophy (CH), ultimately triggering a chronic condition that can culminate in heart failure. 2 The incidence of this disease tends to increase with age. 3 Due to difficulties in treatment and recurrent episodes, it often accompanies severe complications, posing a significant threat to human health. CH and fibrosis are crucial pathological processes in cardiac dysfunction, initially serving as adaptive responses to increased pressure or volume loads but eventually leading to structural and functional deterioration of the heart over time. 4 This disruption of normal myocardial structure and elasticity further exacerbates cardiac function decline. Despite previous efforts in elucidating pathophysiology and developing practical strategies to avoid or reverse CH, satisfying clinical treatments remain elusive. 5 Therefore, investigating new drugs to improve the therapy of CH in patients with pulmonary heart disease is essential.
Robinin (C28H32O15), a flavonoid glycoside isolated from Astragalus species, exhibits potent anti-inflammatory and antioxidant activities by scavenging reactive oxygen species (ROS) and suppressing pro-inflammatory cytokines such as TNF-α and IL-6. 6 In myocardial ischemia-reperfusion models, Robinin attenuates apoptosis via PI3K/Akt signaling. 7 However, its role in modulating hypertrophic pathways, particularly through SIRT1/NF-κB and NLRP3 inflammasome, remains unexplored in pulmonary heart disease-associated CH. This gap underscores the rationale for investigating Robinin’s therapeutic mechanisms in this context.
In recent years, the NLRP3 inflammasome and oxidative stress (OS) have emerged as pivotal players in the pathogenesis of CH and fibrosis. 8 The NLRP3 inflammasome is an intracellular protein, functions as an activator of the Caspase-1 signaling pathway. 9 Numerous studies have indicated the NLRP3 inflammasome can stimulate pro-inflammatory cytokines, notably IL-1β and IL-18 by activating Caspase-1, thus contributing to the development of CH.10,11 OS is an imbalance in the ability of the body to neutralize free radicals using antioxidants, exacerbating myocardial lesions through mechanisms such as damaging cardiomyocytes, promoting inflammatory responses, and inducing apoptosis. 12 Previous research has indicated that OS and the NLRP3 inflammasome has been shown to improve CH and fibrosis through the use of pharmacological agents. 13 However, it remains unclear whether Robinin ameliorates CH by suppressing OS and NLRP3 inflammasome.
SIRT1 has been indicated to play significant roles in various cardiovascular diseases, one of its mechanisms is exerting anti-inflammatory and OS by inhibiting the NF-κB signaling pathway. 14 NF-κB is indeed a critical nuclear transcription factor that plays a pivotal role in regulating cellular inflammation. 15 Furthermore, SIRT1 reduces ROS levels by enhancing mitochondrial function and antioxidant defense through activation of AMPK, PPARα, and PGC-1α, all of which contribute to the suppression of inflammation via inhibition of the NF-κB pathway. 16 However, it is still unclear whether Robinin can exert antioxidant stress effects by regulating the SIRT1/NF-κB pathway.
Therefore, this study aims to investigate whether Robinin alleviates CH and fibrosis by regulating the SIRT1/NF-κB signaling axis, thereby suppressing OS and NLRP3 inflammasome activation. We hypothesize that Robinin ameliorates CH and fibrosis by modulating the SIRT1/NF-κB axis and inhibiting the NLRP3 inflammasome and OS. By systematically evaluating Robinin’s impact on molecular markers of hypertrophy, fibrosis, OS, and the SIRT1/NF-κB pathway, this work seeks to establish its therapeutic potential for CH secondary to pulmonary heart disease.
Method
Experimental animals
SPF male SD rats (n = 8 per group, total N = 48) were selected for the study. Sample size was determined using G*Power 3.1 based on prior studies (effect size = 1.2, α = 0.05, power = 0.8). Rats were randomly assigned to experimental groups (Control, pulmonary heart disease, pulmonary heart disease + Robinin, Sham, etc.) via a computer-generated randomization sequence. Investigators performing echocardiography, tissue sampling, and data analysis were blinded to group assignments. Before starting the formal experiment, the selected rats were acclimated by staying in the experimental environment. The acclimatisation period lasted for 1-2 weeks to reduce disturbing factors in the experiment due to environmental changes. The Animal Ethics Committee of Qiqihar Medical University (Ethics No. QMU-AECC-2024-218) provided ethical approval for all animal trails.
Animal model
Pulmonary hypertension was confirmed by echocardiography and right ventricular systolic pressure (RVSP) measurements. Rats with RVSP 35 mmHg (measured via right heart catheterization under anesthesia) after 4 weeks of 10% CO2 exposure (6 h/day) were included in the study. This approach aligns with prior studies demonstrating that dual stressors better recapitulate human pulmonary heart disease progression.17,18 Rats were anesthetized via intraperitoneal injection of pentobarbital sodium (50 mg/kg body weight; Shanghai Yuanye Biotechnology, China). Postoperative analgesia (meloxicam, 1 mg/kg, subcutaneously) was administered daily for 72 h. Humane endpoints were predefined as severe respiratory distress (>60 breaths/min), inability to eat/drink, or >20% body weight loss; no animals met these criteria. A midline neck incision was made under aseptic conditions. A tracheostomy was performed, and a sterile PE-90 catheter was inserted and secured with 4-0 silk sutures. Rats were then housed in a ventilated chamber (Tecniplast, Italy) with continuous 10% CO2 delivery for 6 h daily over 4 weeks. CH was induced via transverse aortic constriction (TAC): a left thoracotomy was performed, and the aorta was ligated between the brachiocephalic and left carotid arteries using a 6-0 nylon suture tied against a 27-gauge needle. 19 Sham-operated rats underwent identical procedures without aortic ligation. The rats in the pulmonary heart disease + Robinin group were administered Robinin (30 mg/kg, dissolved in 10% DMSO/saline) intragastrically in a volume of 5 mL/kg once daily for 14 consecutive days. This dose was selected based on prior preclinical studies demonstrating efficacy and safety in rodent models of cardiovascular injury. 6 Vehicle control groups received an equal volume of 10% DMSO/saline (5 mL/kg). No adverse effects attributable to DMSO (e.g., mucosal irritation, behavioral changes) were observed at this concentration and volume.
Experimental observation and sampling
Physiological parameters, including heart rate (non-invasive tail-cuff plethysmography), respiratory rate (whole-body plethysmography), and body weight, were recorded daily at 9:00 AM. Behavioral assessments (open-field test for activity levels and forced swim test for stress response) were conducted weekly. Terminal blood and tissue sampling occurred at 4 weeks post-model induction under pentobarbital anesthesia (50 mg/kg). At specific time points, data were collected for subsequent analysis through behavioural tests, blood sample collection and tissue sampling.
Cell culture
H9c2 cardiomyocytes, derived from the BD1X rat cardiac cell line (a subclone of embryonic BD1X rat heart tissue), were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, MA, USA) and 1% penicillin/streptomycin (Invitrogen, CA, USA). The cells were maintained at 37°C in 5% CO2 incubator. H9c2 cells were resuspended at 106 cells/ mL and were seeded on 6-well plates overnight. Subsequently, the cells were serum-starved in serum-free medium for 4 h. Angiotensin II (Ang II) (CAS.NO.: 4474-91-3, Sigma, Shanghai, China), was dissolved in 0.9% saline. Then, 200 nM Ang II was used to process H9c2 cells for 24 h.20,21
Intervention
Robinin (purity ≥ 98%, HPLC-verified; B50946, Shanghai Yuanye Biotechnology Co., Ltd, China) was provided as a lyophilized powder. For in vivo administration, it was dissolved in 10% DMSO (Sigma-Aldrich) diluted in 0.9% saline. A vehicle control group (10% DMSO in saline) was included to account for solvent effects. Oral gavage was selected to simulate clinical oral bioavailability, as prior pharmacokinetic studies indicate Robinin’s stability and absorption via this route. 6 Myocardial cells were handled with Robinin at 10, 20, and 40 µM, or in a culture medium of 3.5 µM for 24 h with SIRT1-IN-1.
Immunofluorescence
The H9c2 cells were incubated overnight at 4°C with the primary antibody against sarcomeric alpha-actinin (1:250; ab137346, Abcam, UK). Next, the cells were incubated with appropriate secondary antibodies for 2 h. 4′,6-diamidino-2-phenylindole (DAPI, Life Technology, Waltham, CA, USA) were used to stain nuclei. A microscope (ZEISS, Germany) was used for fluorescent imaging. No post-acquisition adjustments (e.g., brightness, contrast) were applied to the fluorescent images. The Image-Pro-Plus analytical software was used to quantify the cell surface area. In three different experiments, 50 cells were randomly selected as quantitative indicators, and the average value of the measured data was statistically analyzed. 22
The protein/DNA ratio
H9c2 cells were washed with PBS and scraped in 100 μL lysis buffer. The harvested cell suspension was rotated, and the total protein content was determined by BCA test. The DNA concentration was determined using a DNA quantification kit (Sigma-Aldrich). 23
Real-time fluorescence quantitative PCR (RT-qPCR)
Primer sequences for qRT-PCR analysis.
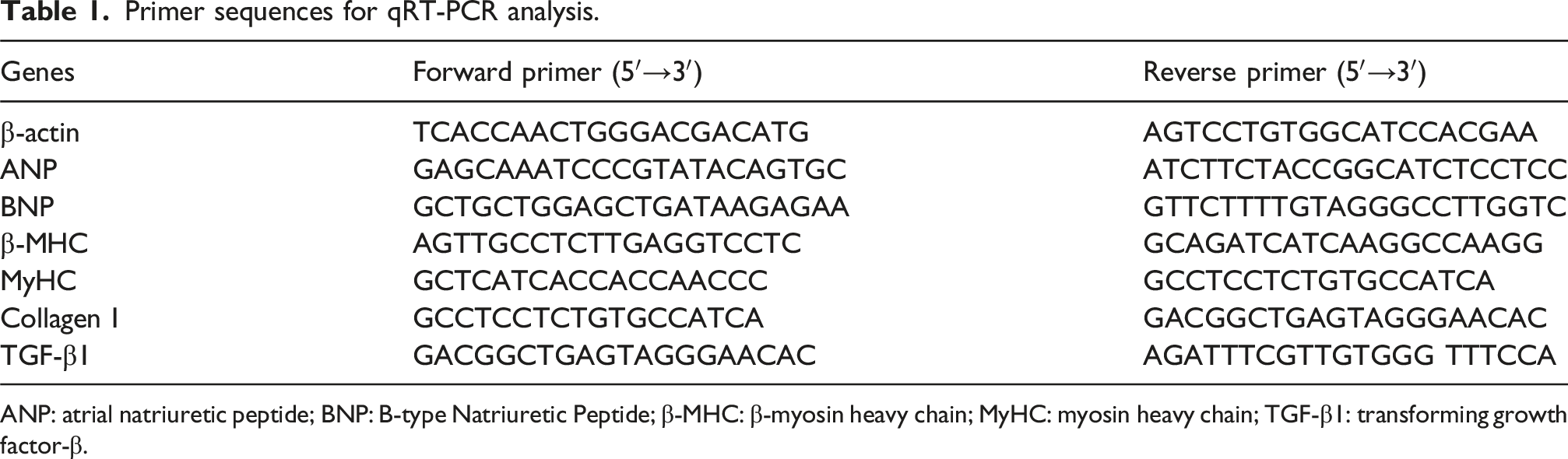
ANP: atrial natriuretic peptide; BNP: B-type Natriuretic Peptide; β-MHC: β-myosin heavy chain; MyHC: myosin heavy chain; TGF-β1: transforming growth factor-β.
ELISA assay
Serum levels of IL-1β, IL-18, ANP, BNP, β-MHC, MyHC, Collagen I, and TGF-β1 were measured using commercial ELISA kits (Elabscience, China) according to the manufacturer’s instructions. Briefly, 100 µL of standards or samples were added to antibody-coated 96-well plates and incubated at 37°C for 90 min. After washing with PBS-T (0.05% Tween-20), 100 µL biotinylated detection antibody was added and incubated for 60 min at 37°C. Plates were washed again, incubated with streptavidin-HRP (1:100 dilution) for 30 min, and developed with TMB substrate (Sigma, USA). The reaction was stopped with 2 M H2SO4, and absorbance was measured at 450 nm using a microplate reader (BioTek Synergy H1, USA). Standard curves were generated for each analyte to calculate concentrations.
Western blot (WB)
Primary antibodies used in Western blot analysis.

OS test
MDA and SOD were selected as OS markers due to their established correlation with CH severity. 25 While MDA and SOD are established surrogates for OS, we acknowledge that direct quantification of reactive oxygen species (e.g., via DCFH-DA flow cytometry or DHE staining) would provide complementary mechanistic insight. Future studies should incorporate these methods to fully characterize Robinin’s antioxidative effects. Our selection of MDA/SOD was based on kit reliability and alignment with prior Robinin studies.6,26
Hematoxylin-Eosin (HE) staining
The cardiac tissues were fixed in 4 % paraformaldehyde and subsequently placed in paraffin. Crosswise sections with a thickness of 5 µm were cut from the embedded tissues and stained with HE (Servicebio, G1003) for routine histological analysis. A high-volume digital holographic scanning system (Pannoramic MIDI, 3DHISTECH, Budapest, Hungary) was used to capture high resolution images of stained sections.
Statistical analysis
The data was analyzed using SPSS 20.0, while GraphPad Prism 9 was utilized for generating visual representations of the experimental data. Statistical significance for comparisons between two groups was assessed using a t-test, while the difference among groups was assessed with one-way ANOVA. The p < .05 was considered statistically significant. In figures, statistical significance is denoted as: *p < .05, **p < .01, *p < .001 versus control; #p < .05, ##p < .01, ###p < .001 versus disease model; &p < .05, &&p < .01, &&&p < .001 versus Robinin-treated group.
Results
Robinin attenuated CH and fibrosis in vitro
H9c2 cardiomyocytes were induced to undergo CH by Ang II, and treated with varying doses of Robinin. As shown in Figure 1(a) and (b), qRT-PCR and ELISA analyses revealed that Ang II significantly upregulated the expression of hypertrophy markers ANP, BNP, and β-MHC, whereas Robinin treatment dose-dependently suppressed these markers (p < .01–.001 vs cardiac hypertrophy group). Similarly, myocardial fibrosis markers MyHC, Collagen I, and TGF-β1 were elevated in the cardiac hypertrophy group but markedly reduced by Robinin in a dose-dependent manner (Figure 1(c) and (d), p < .05–.001). Furthermore, cardiomyocyte surface area and protein/DNA ratio, key indicators of hypertrophy, were significantly increased in the cardiac hypertrophy group (p < .001 vs control). Robinin treatment reversed these changes, with the 40 μM concentration producing the most significant effects (Figure 1(e) and (f), p < .01–.001). These findings collectively demonstrate Robinin’s ability to attenuate CH and fibrosis in vitro. Robinin attenuated CH and fibrosis in vitro. (a) and (b) ANP, BNP, and β-MHC levels. (c) and (d) MyHC, Collagen I, and TGF-β1 levels. (e) The surface area of cardiomyocytes. F: Protein/DNA ratio of cardiomyocytes was calculated using a DNA quantification kit. Data represent mean ± SD. Statistical comparisons: **p < .01, ***p < .001 (vs Control group); #p < .05, ##p < .01, ###p < .001 (vs Cardiac hypertrophy group).
Inhibition of NLRP3 inflammasome and OS by Robinin in vitro
Robinin exhibited potent anti-inflammatory and antioxidant effects. As illustrated in Figure 2(a) and (b), pro-inflammatory cytokines IL-1β and IL-18, as well as NLRP3 inflammasome components (NLRP3, ASC, Caspase1), were significantly elevated in the cardiac hypertrophy group (p < .001 vs control) but suppressed by Robinin in a dose-dependent manner (p < .05–.001 vs cardiac hypertrophy group). OS markers further supported these findings: MDA levels (a lipid peroxidation product) were elevated in the cardiac hypertrophy group (p < .001 vs control), while SOD activity and antioxidant proteins HO-1/Nrf2 were reduced. Robinin treatment normalized MDA levels, enhanced SOD activity, and upregulated HO-1/Nrf2 expression (Figure 2(c)–(f), p < .05–.001). These results confirm Robinin’s dual role in inhibiting NLRP3 inflammasome activation and mitigating OS. Inhibition of NLRP3 inflammasome and OS by Robinin in vitro. (a) IL-1β and IL-18 levels. (b) NLRP3 inflammasome level. (c) HO-1, p-Nrf2 and Nrf2 levels. (d) p-Nrf2/Nrf2. E: MDA level. F: SOD level. Data represent mean ± SD. Statistical comparisons: ***p < .001 (vs Control group); #p < .05, ##p < .01, ###p < .001 (vs Cardiac hypertrophy group).
Robinin regulated SIRT1/NF-κB pathway in vitro
Previous research has indicated SIRT1 may interact with NF-kB signaling pathway to alleviate CH.
27
To elucidate the molecular mechanism, we investigated the SIRT1/NF-κB axis. CH induction downregulated SIRT1 expression and activated the NF-κB pathway (increased p-NF-κB/NF-κB ratio, p < .001 vs control). Robinin treatment dose-dependently restored SIRT1 levels and suppressed NF-κB phosphorylation (Figure 3(a) and (b), p < .05–.001 vs cardiac hypertrophy group), suggesting SIRT1-mediated NF-κB inactivation contributes to Robinin’s therapeutic effects. Robinin regulated SIRT1/NF-κB pathway in vitro. (a) SIRT1, p-NF-κB and NF-κB levels. (b) p-NF-κB/NF-κB. Representative Western blot images are shown without selective adjustments. Data represent mean ± SD. Statistical comparisons: *p < .05, ***p < .001 (vs Control group); #p < .05, ##p < .01, ###p < .001 (vs Cardiac hypertrophy group).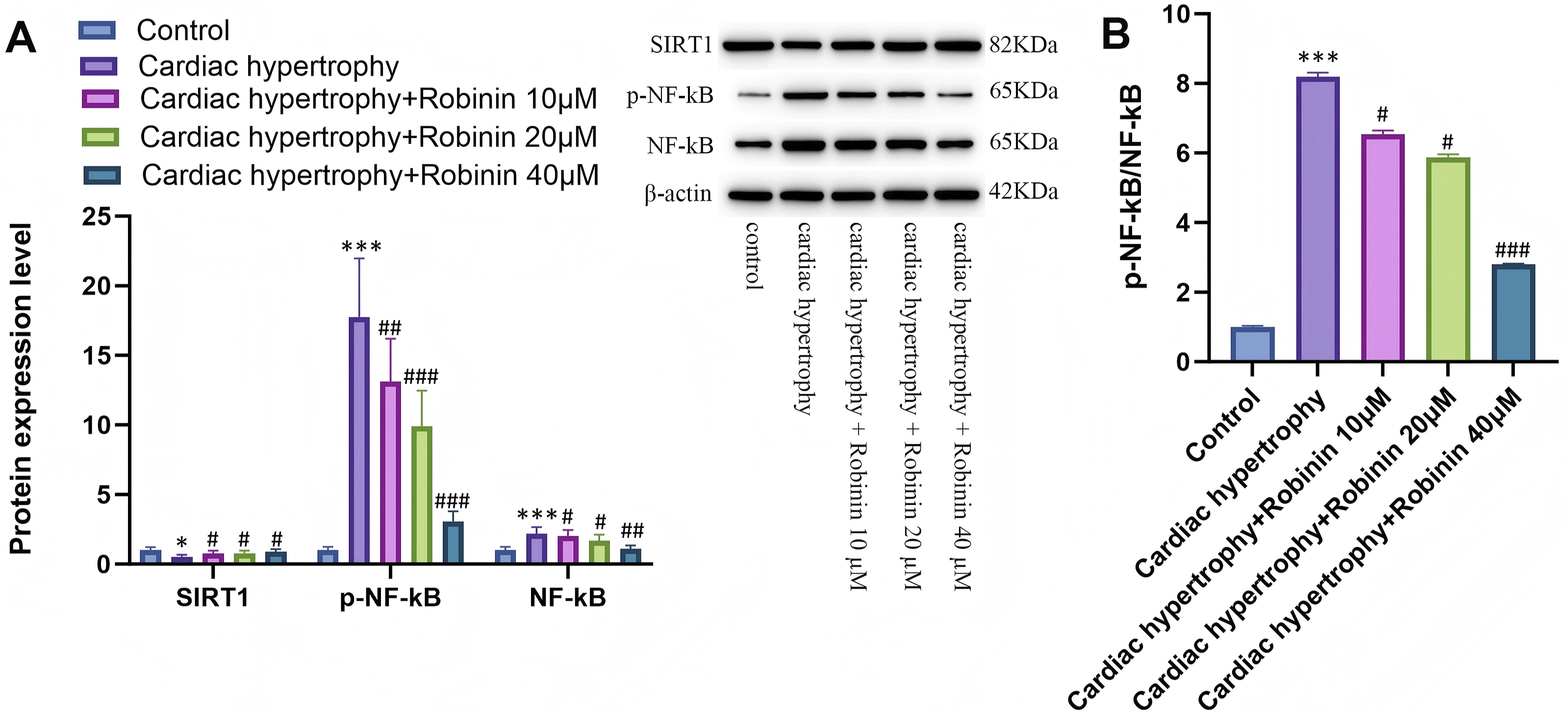
SIRT1 inhibition attenuated Robinin’s protection against CH in vitro
Pharmacological inhibition of SIRT1 (using SIRT1-IN-1) abolished Robinin’s protective effects. Co-treatment with SIRT1-IN-1 reversed the suppression of ANP, BNP, β-MHC, MyHC, Collagen I, and TGF-β1 by Robinin (Figure 4(a)–(d), p < .05–.001 vs Robinin-alone group). Similarly, SIRT1 inhibition exacerbated cardiomyocyte surface area enlargement and protein/DNA ratio (Figure 4(e) and (f), p < .01–.001), confirming SIRT1 is critical for Robinin’s anti-hypertrophic action. SIRT1 inhibition attenuated Robinin’s protection against CH in vitro (a) and (b) ANP, BNP, and β-MHC levels. (c) and (d) MyHC, Collagen I, and TGF-β1 levels. (e) The surface area of cardiomyocytes. (f) The protein/DNA ratio of cardiomyocytes. Data represent mean ± SD; n = 8 rats per group. Statistical comparisons: *p < .05, **P < .01, ***p < .001 (vs Control group); #p < .05, ##p < .01, ###p < .001 (vs Cardiac hypertrophy group); && p < .01, &&& p < .001 (vs Cardiac hypertrophy + Robinin group).
SIRT1 inhibition reversed Robinin’s suppression of NLRP3 inflammasome and OS
SIRT1 inhibition also negated Robinin’s anti-inflammatory and antioxidant effects. IL-1β, IL-18, NLRP3, ASC, and Caspase1 levels, which were suppressed by Robinin, rebounded upon SIRT1-IN-1 co-treatment (Figure 5(a) and (b), p < .05–.001). Similarly, SIRT1 blockade reversed Robinin’s normalization of MDA/SOD and HO-1/Nrf2 levels (Figure 5(c) and (f), p < .05–.001), further linking SIRT1 to Robinin’s modulation of OS and inflammasome activity. Inhibiting SIRT1 attenuated Robinin’s inhibition of NLRP3 inflammasome and OS in vitro. (a) IL-1β and IL-18 levels. (b) NLRP3, ASC and Caspase1 levels. (c) HO-1, p-Nrf2 and Nrf2 levels. (d) p-Nrf2/Nrf2. (e) MDA level. (f) SOD level. Data represent mean ± SD; n = 8 rats per group. Statistical comparisons: *p < .05, **p < .01, ***p < .001 (vs Control group); #p < .05, ##V < .01, ###p < .001 (vs Cardiac hypertrophy group); & p < .05, && p < .01 (vs Cardiac hypertrophy + Robinin group).
Inhibiting SIRT1 reduced Robinin’s regulation on SIRT1/NF-κB pathway induced by pulmonary heart disease in vitro
Compared with cardiac hypertrophy group, the cardiac hypertrophy + SIRT1-IN-1 group exhibited downregulation of SIRT1 and upregulation of p-NF-κB and NF-κB (Figure 6A, B, p < .05–.01). Concurrently, compared to the cardiac hypertrophy + Robinin group, the addition of SIRT1-IN-1 led to downregulation of SIRT1 and upregulation of NF-κB. These findings indicate that inhibiting SIRT1 can reduce Robinin’s regulation on NF-κB pathway. Inhibiting SIRT1 reduced Robinin’s regulation on SIRT1/NF-κB pathway induced by pulmonary heart disease in vitro. Representative Western blot images are shown without selective adjustments. Data represent mean ± SD; n = 8 rats per group. Statistical comparisons: *p < .05, **p < .01, ***p < .001 (vs Control group); #p < .05, ##p < .01 (vs Cardiac hypertrophy group); & p < .05, && p < .01 (vs Cardiac hypertrophy + Robinin group).
Robinin alleviated CH in vivo
In a rat model of pulmonary heart disease, Robinin (30 mg/kg) significantly ameliorated CH and fibrosis. Elevated levels of ANP, BNP, β-MHC, MyHC, Collagen I, and TGF-β1 in pulmonary heart disease rats were reduced by Robinin (Figure 7(a)–(d), p < .05–.001). NLRP3 inflammasome activation and inflammatory cytokines (IL-1β, IL-18) were suppressed, while HO-1/Nrf2 expression and SIRT1 levels were restored (Figure 7(e)–(l), p < .05–.001). Histopathological analysis revealed reduced cardiomyocyte hypertrophy and fibrosis in Robinin-treated rats (Figure 7g), corroborated by a normalized heart weight/body weight ratio (Figure 7h, p < .001). These in vivo results align with in vitro findings, solidifying Robinin’s therapeutic potential. Robinin alleviated CH in vivo. (a) and (b) ANP, BNP, and β-MHC levels. (c) and (d)MyHC, Collagen I, and TGF-β1 levels. (e) IL-1β and IL-18 levels. (f) NLRP3 inflammasome level. (g) Examination of rat heart histopathology. (h) Heart weight (HW) /body weight (BW). (i) SIRT1, p-NF-κB and NF-κB levels. (j) p-NF-κB/NF-κB. (k) HO-1, p-Nrf2 and Nrf2 levels. (l) p-Nrf2/Nrf2. Representative Western blot images are shown without selective adjustments. Data represent mean ± SD; n = 8 rats per group. Statistical comparisons: *p < .05, **p < .01, ***p < .001 (vs Control group); #p < .05, ##p < .01, ###p < .001 (vs Pulmonary heart disease group).
Discussion
CH is a multifaceted cardiovascular disease that progresses to heart failure, and persistent CH is irreversible. 28 Thus, studying the pathogenesis of CH is of significant importance. Traditional anti-hypertrophic drugs, such as beta-blockers, ACE inhibitors, and ARBs, primarily work by modulating the neuroendocrine system, decreasing cardiac load and pressure, indirectly influencing myocardial remodeling. 29 Although widely used clinically, these medications have limited efficacy for some patients and come with certain side effects. In this study, Robinin reduced the expression of key markers during CH and fibrosis, indicating its significant therapeutic effects in reducing CH and fibrosis.
Mechanistic insights: SIRT1/NF-κB axis
Robinin, a flavonoid glycoside derived from Astragalus falcatus, demonstrates multifaceted cardioprotective properties. Our findings indicate that Robinin’s cardioprotection involves SIRT1-mediated NF-κB inactivation and Nrf2 activation, leading to reduced OS and inflammation. This is consistent with previous reports showing Robinin’s ability to mitigate endoplasmic reticulum stress and apoptosis in H9c2 cells, and enhance recovery from myocardial ischemia-reperfusion injury via PI3K/Akt signaling. 6 Robinin enhances the recovery form myocardial ischemia-reperfusion injury by activating antioxidant enzymes and inhibiting apoptosis.7,30 Its strong antioxidant properties help scavenge free radicals, reducing OS damage to the cardiovascular system, while also diminishing inflammatory responses and lowering the risk of cardiovascular events. 31
While our data strongly support SIRT1/NF-κB as a key axis for Robinin’s effects, SIRT1 is a multifunctional regulator that interacts with other pathways, such as AMPK/PGC-1α and PI3K/Akt. 32 For instance, SIRT1 activation can enhance mitochondrial biogenesis via AMPK/PGC-1α, indirectly reducing OS. 33 Similarly, Robinin’s reported PI3K/Akt-mediated antioxidant effects may synergize with SIRT1/NF-κB modulation. Further studies are needed to delineate hierarchical or parallel contributions of these pathways. 6
Comparison with other flavonoids
Unlike Acacetin, which primarily utilizes the SIRT1/AMPK/PGC-1α pathway for anti-hypertrophic action, 34 Robinin’s protection in pulmonary heart disease-induced CH is mediated predominantly through SIRT1-dependent suppression of NF-κB and its downstream effectors NLRP3 inflammasome and OS. Similarly, while Puerarin reduces CH via modulation of the ERK1-2/p38/NF-κB pathway, 31 Robinin’s mechanism centers on direct SIRT1 upregulation leading to NF-κB inactivation, representing a distinct therapeutic node.
This SIRT1/NF-κB engagement is central to Robinin’s efficacy. Consistent with established roles where SIRT1 activation inhibits CH and regulates NF-κB activity to mitigate inflammation and OS,35,36 our findings demonstrate Robinin dose-dependently upregulated SIRT1 and inactivated NF-κB. Crucially, pharmacological inhibition of SIRT1 abolished Robinin’s protection against hypertrophy, fibrosis, inflammation, and OS, confirming SIRT1’s indispensable role and the pathway’s specificity in this context. This result suggests that SIRT1 activity is essential for Robinin’s anti-CH and anti-fibrosis effects, and Robinin mediates cardioprotection via the SIRT1/NF-κB pathway.
Clinical value
Robinin has indicated its potential in treating CH and fibrosis through inhibition of NLRP3 inflammasome and reduction of OS. This discovery not only provides new insights into the therapeutic mechanisms of Robinin but also opens avenues for developing novel therapies targeting the NLRP3 inflammasome and OS. Other natural products or synthetic drugs might share similar mechanisms of action, inhibiting the activation of the NLRP3 inflammasome or modulating OS, making them promising candidates for cardiovascular disease treatment. Given the complexity of cardiovascular disease etiology and individual variability, the application of Robinin may require personalization based on an individual’s genetic background, environmental factors, and lifestyle, leading to more precise treatment strategies.
Limitations and future directions
While Robinin has indicated promising therapeutic effects, further research is needed to investigate its pharmacokinetic properties, dose-response relationships, and long-term safety to ensure its safety and efficacy as a therapeutic agent for cardiovascular diseases. While MDA and SOD are established markers of OS, direct ROS quantification (e.g., DCFDA staining) or reactive nitrogen species assessments were not performed in this study. Future investigations should incorporate these methods to further validate Robinin’s antioxidative effects. Nevertheless, our findings provide a scientific foundation for Robinin as a potential therapeutic agent; however, its feasibility in clinical trials still needs to be substantiated. Additionally, the absence of direct ROS measurements (e.g., DCFH-DA) represents a methodological limitation; future work should validate our OS findings using real-time ROS detection assays. Future research should also address the need for personalized approaches, given the complexity of cardiovascular disease etiology and individual variability in genetic backgrounds, environmental exposures, and lifestyle factors.
Conclusion
Our results confirm that Robinin alleviates CH through modulation of the SIRT1/NF-κB pathway by inhibiting OS and NLRP3 activation, demonstrating its therapeutic potential for CH secondary to pulmonary heart disease. This discovery provides a scientific basis for Robinin as a potential therapeutic agent; its feasibility in clinical application remains to be established.
Footnotes
Ethical approval
The Animal Ethics Committee of Qiqihar Medical University (Ethics No. QMU-AECC-2024-218) provided ethical approval for all animal trails.
Author’s contributions
Chao Wang designed the study. Yubao Liu provided administrative support. Chao Wang, Yubao Liu and Lei Qin provided materials. Chao Wang, Xinrui Zhang collected the data. Chao Wang, Yubao Liu and Lei Qin analyzed the data. All authors wrote the manuscript. All authors reviewed the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Qiqihar Science and Technology Program Joint Guidance Project (LHYD-202047).
Declarations conflicts of interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.