
Abstract
Purpose
Lung adenocarcinoma (LUAD) is the most prevalent subtype of non-small cell lung cancer and a leading cause of cancer-related mortality worldwide. This study investigates the role of Ring Finger Protein 216 (RNF216) in LUAD progression.
Methods
RNF216 expression was evaluated in LUAD tissues and cells. Functional assays evaluated cell viability, migration, invasion, and ferroptosis in vitro. Mechanistic investigations defined RNF216’s role in regulating p53 ubiquitination and stability. In vivo, xenograft models evaluated tumor growth and ferroptosis.
Results
RNF216 was markedly overexpressed in LUAD tissues and cell lines. Functional studies demonstrated that silencing RNF216 suppressed LUAD cell proliferation, migration, and invasion while inducing ferroptosis, characterized by increased reactive oxygen species (ROS), lipid peroxidation (LPO), and intracellular Fe2+ accumulation. Mechanistically, RNF216 knockdown stabilized p53 by reducing its ubiquitination, thereby promoting ferroptosis. These findings were corroborated in vivo, where RNF216 silencing significantly inhibited tumor growth and enhanced ferroptosis in xenograft models.
Conclusions
Our results establish RNF216 as a pivotal oncogenic driver that accelerates LUAD progression by suppressing ferroptosis through p53 ubiquitination. Targeting RNF216 may represent a promising therapeutic strategy to induce ferroptosis and combat LUAD.
Introduction
Lung cancer is the most commonly diagnosed cancer globally, with non-small cell lung cancer (NSCLC) represents about 85% of cases. 1 Among NSCLC subtypes, lung adenocarcinoma (LUAD) demonstrates particularly high incidence and mortality rates. 2 The pervasiveness of lung cancer underscores the urgent need for an in-depth understanding of the molecular mechanisms driving its progression, with a particular emphasis on identifying new therapeutic targets.
Ferroptosis, a form of programmed cell death characterized by iron-dependent lipid peroxidation, has recently gained prominence as a crucial process in the regulation of tumor growth, including LUAD.3,4 Unlike other forms of cell death, such as apoptosis or necrosis, ferroptosis is driven by the accumulation of lethal lipid peroxides, making it uniquely dependent on iron metabolism and redox homeostasis. 5 Inducing ferroptosis in cancer cells has emerged as a promising therapeutic strategy, offering potential advantages in overcoming drug resistance and targeting aggressive tumor phenotypes. 6
The tumor suppressor p53 orchestrates diverse cellular responses, including apoptosis, cell cycle arrest, and DNA repair in response to cellular stresses. 7 Beyond its canonical functions, p53 is implicated in the regulation of iron metabolism, lipid peroxidation, and antioxidant pathways, thereby exerting significant influence over ferroptosis. 8 Recent studies suggest that p53 modulates ferroptosis via the SLC7A11/GPX4 axis in lung cancer. 9 Moreover, p53 activity is intricately controlled by ubiquitination, a post-translational modification that targets proteins for degradation or functional modulation. 10 This ubiquitination process is increasingly recognized as a critical factor in determining the fate of p53 and its role in ferroptosis regulation in human cancers.11,12 However, the interplay between p53 ubiquitination and ferroptosis in LUAD remains poorly understood.
Ring finger protein 216 (RNF216, also known as TRIAD3) is a multifunctional E3 ubiquitin ligase encoded by the RNF216 gene located at 7p22.1. 13 The RNF216 protein consists of approximately 866 amino acids. 14 RNF216 is ubiquitously expressed across various tissues, most predominantly in the brain. 15 RNF216 plays a key role in multiple cellular processes, including cell growth, 16 migration, 17 apoptosis, 18 autophagy, 19 and inflammation. 20 Prior studies have shown that RNF216 is aberrantly expressed and plays different roles in human cancers. 21 For instance, RNF216 is downregulated in ovarian cancer and inhibits cancer development. 16 On the contrary, RNF216 is upregulated in colorectal cancer and promotes tumor growth and metastasis by inhibiting BECN1-dependent autophagy. 22 Notably, RNF216 can attenuate radiation-induced apoptosis and DNA damage in glioblastoma via induction of p53 ubiquitination and degradation, 23 suggesting RNF216 may modulate cell ferroptosis via p53. In LUAD, RNF216 is upregulated and correlates with poor prognosis, 24 underscoring its potential significance in LUAD progression. We thus proposed that RNF216 may serve as a crucial mediator in the regulation of ferroptosis in LUAD, potentially through its interaction with p53.
This study aims to investigate the role of RNF216 in LUAD progression by focusing on its interaction with p53. By exploring how RNF216 modulates ferroptosis through p53 ubiquitination, this research seeks to provide novel insights into the molecular network underlying LUAD and identify potential therapeutic targets to enhance ferroptosis-based cancer treatments.
Materials and methods
Clinical samples
This research was conducted ethically in accordance with the World Medical Association Declaration of Helsinki. The present study received approval from the Institutional Ethics Committee of The Second Affiliated Hospital of Jiaxing University. Tumor tissues and corresponding adjacent normal lung tissues were procured from 45 patients with LUAD who underwent surgical resection at The Second Affiliated Hospital of Jiaxing University, all of whom had not received prior chemotherapy or radiotherapy. Written informed consent was obtained from each participant. All samples were promptly frozen in liquid nitrogen following their excision.
Cell culture and transfection
Human LUAD cell lines (A-549, PC-9, and NCI-H1975) and normal bronchial epithelial cell line (Beas-2B) were obtained from BeNa Culture Collection (BNCC, China). A-549, PC-9, and NCI-H1975 cells were maintained in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin under humidified conditions (37°C; 5% CO2; 95% relative humidity). 25 Beas-2 B cells were maintained in DMEM-H medium supplemented with 10% FBS and 1% penicillin/streptomycin under humidified conditions (37°C; 5% CO2; 95% relative humidity). 26
Short hairpin RNA (shRNA) targeting RNF216 (sh-RNF216) and the corresponding negative control (sh-NC) were designed and constructed by GenePharma (China). These shRNAs were cloned into pLKO.1 shRNA lentiviral vector (Addgene) and transfected into HEK-293T cells together with psPAX2 and pMD2.G using Lipofectamine® 3000 reagent and cultured for 48 h. Next, lentivirus particles containing sh-RNF216 or sh-NC were collected and filtered to infect LUAD cells (A-549 and PC-9). 48 h after infection, stable clones were selected by puromycin treatment (2 μg/mL). 27
RT-qPCR
Total RNA was isolated from tissue samples or cells using TRIzol Reagent (Invitrogen) and subsequently reverse transcribed into cDNA with a PrimeScript RT reagent Kit (Takara, China) as per the manufacturer’s instructions. Then, qPCR was performed as previously described. 28
Western blotting
Cellular proteins were extracted using RIPA buffer and subjected to separation via 10% SDS-PAGE. The proteins were then transferred to PVDF membranes. The membranes were blocked with 5% skim milk, incubated with primary antibodies at 4°C overnight, and subsequently incubated with secondary antibodies at room temperature for 1 h. Protein bands were detected with an electrochemiluminescence (ECL) Kit (Amersham Biosciences). 29
CCK-8 assay
Cell viability of LUAD cells was evaluated using CCK-8 assay. 30 Cells were plated in 96-well plates (5 × 103/well). After incubation for the specified duration, 10 μL of CCK-8 reagent was added to each well. Optical density was then measured at 450 nm under an automated microplate reader.
Colony-formation assay
Colony-forming assays were performed to determine the clonogenic activity of LUAD cells. 31 Briefly, LUAD cells were seeded in 6-well plates and cultured till visible colonies formed. The colonies were then washed with PBS, fixed with methanol, and stained with Giemsa solution.
Transwell assay
Cell invasion and migration were examined using Transwell inserts (8-µm pore size; Corning Inc.) with or without Matrigel coating (BD Biosciences, USA). 32 LUAD cells were suspended in serum-free DMEM and placed in the upper chamber and allowed to migrate or invade the lower chamber (90% DMEM; 10% FBS). After 48 h, non-migratory or non-invasive cells on the upper surface of the filters were removed. The migrated or invaded cells were subsequently stained with 0.1% crystal violet and quantified under a microscope.
Detection of lipid peroxidation (LPO) and intracellular reactive oxygen species (ROS)
Intracellular ROS and LPO were evaluated using DCFH-DA (Beyotime) and BODIPY™ 581/591 C11 (Invitrogen) Beyotime), as previously described. 33
Intracellular ferrous iron (Fe2+) measurement
Fe2+ level in LUAD cells was determined using the Iron Assay Kit (Beyotime), as previously described. 34
Detection of malondialdehyde (MDA) level and glutathione (GSH)/oxidized glutathione (GSSG) ratio
MDA level and GSH/GSSG ratio in LUAD cells were determined using specific commercial kits (Nanjing Jiancheng Biotechnology Co. Ltd, China), as previously described. 35
Co-immunoprecipitation (Co-IP) and ubiquitination assays
Co-IP and ubiquitination assays were performed as previously described. 36 For the Co-IP assay, whole cell lysates were extracted and incubated with anti-p53, anti-RNF216, and IgG for 4 h. Subsequently, the precipitated proteins were captured using Protein A/G PLUS-Agarose and analyzed using western blotting with specified antibodies.
For ubiquitination assay, cell lysates were prepared and subjected to immunoprecipitation with anti-p53 antibody. To assess ubiquitinated p53 levels, the precipitated proteins were analyzed by Western blotting with an anti-ubiquitin antibody.
Animal study
Male BALB/c-nude mice (1–2 months old, 20 ± 2 g) maintained under specific pathogen-free conditions were used. As previously described, 37 each mouse was injected with A-549 cells stably transfected with sh-RNF216 (n = 3) or sh-NC (n = 3) (5 × 106 cells). Tumor growth was monitored every 3 days, with tumor volume calculated using the formula: volume (mm3) = longer diameter × shorter diameter 2 × 0.5. All mice were sacrificed to harvest xenografted tumors 3 weeks after injection. Protocols for animal experiments were approved by the Animal Experimental Ethics Committee of The Second Affiliated Hospital of Jiaxing University.
Immunohistochemical (IHC) staining
As previously described, 38 IHC staining was performed to evaluate Ki-67 and GPX4 expression in xenografted tumors.
Statistical analyses
All the statistical analyses were conducted using GraphPad Prism 6.0 (GraphPad, USA). Data were expressed as means ± SD. Comparisons between groups were performed using Student’s t-test or one-way ANOVA, as appropriate. A p-value < .05 was considered indicative of statistical significance. The reproducibility of the results was confirmed in at least three independent experiments.
Results
RNF216 is highly expressed in LUAD
LUAD tissues and adjacent normal tissues were collected from 45 patients with LUAD. RT-qPCR results confirmed that RNF216 is significantly upregulated in LUAD tissues relative to adjacent normal tissues (Figure 1(a)). Additionally, both mRNA and protein levels of RNF216 were assessed in LUAD cell lines (A-549, PC-9, and NCI-H1975) and a normal bronchial epithelial cell line (Beas-2B) using RT-qPCR and Western blotting. As illustrated in Figure 1(b) and (c), RNF216 was markedly elevated in LUAD cells compared to normal cells. Due to particularly high expression levels of RNF216 in A-549 and PC-9 cells, these lines were selected for further experimental analysis. Taken together, RNF216 was upregulated in LUAD cells. RNF216 is highly expressed in LUAD. (a) RT-qPCR for RNF216 mRNA expression in 45 pairs of LUAD tissues and corresponding peritumor tissues. (b and c) RT-qPCR and western blotting for RNF216 mRNA and protein levels in LUAD cells (A-549, PC-9, and NCI-H1975) and normal cells (Beas-2B). *p < .05; **p < .01.
RNF216 deletion inhibits LUAD cell growth, migration, and invasion
To elucidate the role of RNF216 in LUAD in vitro, RNF216 was knocked down in A-549 and PC-9 cells via sh-RNF216 transfection. Subsequent RT-qPCR and Western blot analyses confirmed reduced RNF216 mRNA and protein levels in these cells (Figure 2(a) and (b)). Compared to the sh-NC group, RNF216 silencing led to decreased cell viability at 48 and 72 h and a reduction in colony formation (Figure 2(c) and (d)). Additionally, RNF216 knockdown significantly impaired cell migration and invasion in A-549 and PC-9 cells (Figure 2(e) and (f)). These findings suggest that RNF216 deletion suppressed LUAD cell proliferation, migration, and invasion. RNF216 deletion inhibits LUAD cell growth, migration, and invasion. (a) RT-qPCR for RNF216 mRNA expression in A-549 and PC-9 cells transfected with sh-NC or sh-RNF216. (b) Western blotting for RNF216 protein expression. (c) CCK-8 assay for cell viability of A-549 and PC-9 cells. (d) Colony formation assay for colony formation efficiency in A-549 and PC-9 cells. (e and f) Transwell assays for migration and invasion of A-549 and PC-9 cells. *p < .05; **p < .01.
RNF216 inhibition promotes ferroptosis in LUAD cells
Ferroptosis is implicated in LUAD progression and treatment.
39
The mRNA expression of ferroptosis marker genes, including SLC7A11, GPX4, FTH1, and ACSL4, were detected in LUAD and normal tissues by RT-qPCR. Consistent with previous studies, we found that SLC7A11, GPX4, and FTH1 expression was upregulated, while ACSL4 expression was downregulated in LUAD tissues compared to adjacent normal tissues (Figure 3(a)–(d)), indicating that ferroptosis may be inhibited in LUAD. To assess the impact of RNF216 on ferroptosis in LUAD in vitro, ferroptosis-related markers were analyzed in A-549 and PC-9 cells. RNF216 depletion significantly increased intracellular ROS, LPO, and Fe2+ levels, compared with the sh-NC group (Figure 3(e)–(g)). In addition, the expression levels of GPX4 and FTH1, which are critical for ferroptosis regulation,
6
were reduced upon RNF216 silencing (Figure 3(h)). As shown in Figure 3(i) and (j), RNF216 knockdown increased the MDA level but reduced the GSH/GSSG ratio. Therefore, RNF216 knockdown promoted ferroptosis in LUAD cells. RNF216 inhibition promotes ferroptosis in LUAD cells. (a–d) SLC7A11, GPX4, FTH1, and ACSL4 mRNA expression in 45 pairs of LUAD tissues and corresponding peritumor tissues. (e–g) ROS, LOP, and Fe2+ levels were determined by specific kits. (H) The expression of ferroptosis-related genes (FTH1 and GPX4) was determined by RT-qPCR. (i and j) MDA level and GSH/GSSG ratio were determined by the appropriate kits. *p < .05; **p < .01.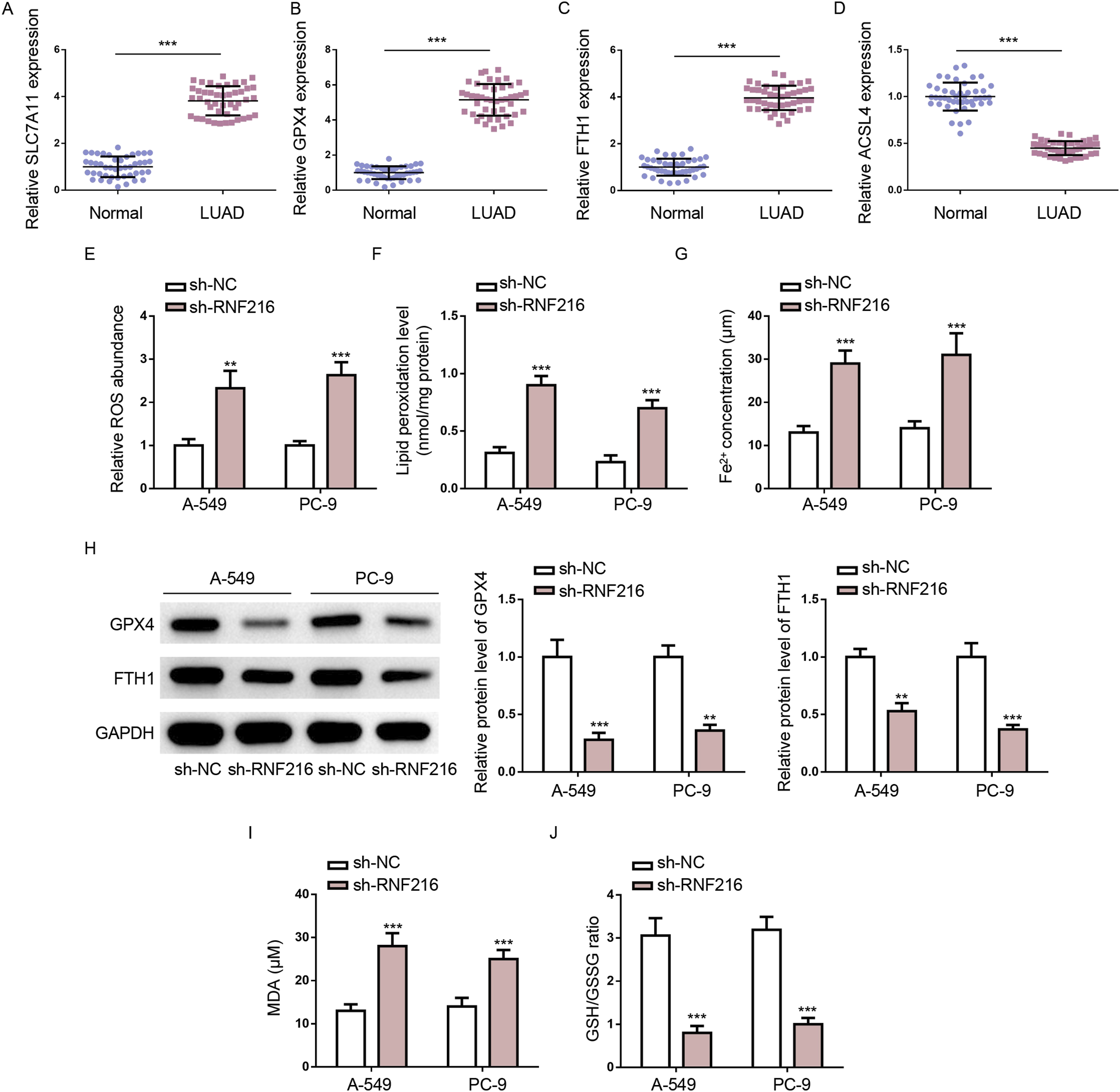
RNF216 silencing inhibits malignant phenotypes of LUAD cells by promoting ferroptosis
To determine if RNF216 regulates LUAD progression through ferroptosis, A-549 and PC-9 cells were treated with the ferroptosis inhibitor ferrostatin-1 (Fer-1). Fer-1 treatment mitigated the effects of RNF216 silencing on cell viability and proliferation (Figure 4(a) and (b)) and increased cell migration and invasion (Figure 4(c) and (d)) compared to the sh-RNF216 group. Therefore, Fer-1 treatment counteracted the inhibitory effects of RNF216 silencing on LUAD cell growth and invasiveness. RNF216 silencing inhibits malignant phenotypes of LUAD cells by promoting ferroptosis. (a) Cell viability of A-549 and PC-9 cells from sh-NC, sh-RNF216, and sh-RNF216+Fer-1 groups. (b) Colony formation efficiency of A-549 and PC-9 cells from each group. (c and d) A-549 and PC-9 cell migration and invasion. *p < .05; **p < .01; ***p < .001.
The role of RNF216 in LUAD tumor growth and ferroptosis in vivo
To evaluate the role of RNF216 in vivo, xenograft models were established using A-549 cells transfected with either sh-RNF216 (n = 3) or sh-NC (n = 3). All nude mice developed xenograft tumors at the injection site, and the tumors were harvested 3 weeks after injection. All tumors from the sh-RNF216 group exhibited significant growth inhibition compared to those from the sh-NC group, consistent with the in vitro findings (Figure 5(a)–(c)). RT-qPCR analysis revealed a significant reduction in RNF216 expression in tumors from the sh-RNF216 group (Figure 5(d)). Immunohistochemical (IHC) analysis showed decreased expression of Ki-67 and GPX4 in tumors with RNF216 silencing (Figure 5(e)). These results supported the hypothesis that RNF216 promoted tumor growth and suppresses ferroptosis in LUAD in vivo. The role of RNF216 in LUAD tumor growth and ferroptosis in vivo. (a–c) Tumor size, weight, and volume of xenograft models were established using A-549 cells stably transfected with sh-RNF216 (n = 3) or sh-NC (n = 3). (d) RT-qPCR for RNF216 mRNA expression in xenograft tumors. (e) IHC for Ki-67 and GPX4 expression in xenograft tumors. Reproducibility of the results was confirmed in at least three independent experiments. *p < .05; **p < .01.
RNF216 depletion increases p53 expression by inhibiting p53 ubiquitination and degradation in LUAD cells
Studies have shown that p53 ubiquitination and degradation are deeply involved in LUAD progression.
36
Interestingly, RNF216 can reduce the stability of p53 protein by promoting p53 ubiquitination.
23
Therefore, we confirmed the interaction between RNF216 and p53 in LUAD cells. Co-IP results indicated the interaction of RNF216 and p53 in A-549 and PC-9 cells (Figure 6(a)). Next, CHX chase assays demonstrated that RNF216 knockdown decelerated p53 degradation in A-549 and PC-9 cells (Figure 6(b)). In addition, RNF216 knockdown upregulated p53 protein levels in A-549 and PC-9 cells, which was further enhanced by MG132 treatment (Figure 6(c)). Furthermore, RNF216 knockdown decreased p53 ubiquitination in A-549 and PC-9 cells (Figure 6(d)). Therefore, RNF216 reduced p53 stability in LUAD cells by promoting p53 ubiquitination. RNF216 depletion increases p53 expression by inhibiting p53 ubiquitination and degradation in LUAD cells. (a) Co-IP for interaction between RNF216 and p53 in A-549 and PC-9 cells. (b) Western blotting for p53 expression after CHX treatment after indicated time. (c) Western blotting for p53 protein level in A-549 and PC-9 cells from sh-NC, sh-NC + MG132, sh-RNF216, and sh-RNF216+MG132 groups. (d) p53 ubiquitin level in A-549 cells from sh-NC and sh-RNF216 groups. **p < .01; ***p < .001.
Inhibition of p53 blocks the impacts of RNF216 knockdown
To clarify the role of p53 in RNF216-mediated promoting effects of malignant phenotypes of LUAD cells, a p53 inhibitor, pifithrin-α (PFT-α), was used to treat LUAD cells. PFT-α reversed the increase in p53 protein levels induced by RNF216 knockdown (Figure 7(a)) and mitigated the inhibitory effects of RNF216 silencing on LUAD cell growth (Figure 7(b) and (c)). Moreover, p53 inhibition also blocked the suppressive effect of RNF216 silencing on LUAD cell migration and invasion (Figure 7(d) and (e)). Notably, PFT-α intervention also reversed the effects of RNF216 silencing on ferroptosis (Figure 8(a)–(g)). These results suggested that RNF216 facilitates LUAD malignancy and suppresses ferroptosis by promoting p53 ubiquitination and degradation. Inhibition of p53 blocks the effects of RNF216 knockdown on LUAD cell growth, migration, and invasion. (a) p53 protein level in A-549 and PC-9 cells from sh-NC, sh-RNF216, and sh-RNF216+PFT-α groups. (b) Cell viability of A-549 and PC-9 cells from each group. (c) Colony formation efficiency of A-549 and PC-9 cells from each group. (d and e) A-549 and PC-9 cell migration and invasion. **p < .01; ***p < .001. Inhibition of p53 blocks the impacts of RNF216 knockdown. (a–c) ROS, LOP, and Fe2+ levels in A-549 and PC-9 cells from sh-NC, sh-RNF216, and sh-RNF216+PFT-α groups. (d) FTH1 and GPX4 expression levels in A-549 and PC-9 cells from each group. (e and f) MDA level and GSH/GSSG ratio in A-549 and PC-9 cells from each group. **p < .01; ***p < .001.

Discussion
RNF216 is deeply involved in various human cancers.16,23,40 A preceding study unveiled the upregulation of RNF216 in LUAD, establishing a positive correlation with an adverse prognosis, 24 indicating RNF216 might play an oncogenic role in LUAD. This study further elucidated the role of RNF216 in regulating LUAD cell functions and the underlying mechanisms. RNF216 was upregulated in LUAD tissue samples and cells. In addition, RNF216 knockdown significantly inhibited the viability, colony formation capability, and migrative and invasive abilities of LUAD cells.
Ferroptosis is an iron-dependent form of regulated cell death characterized by LPO accumulation. 41 Ferroptosis dysregulation plays a vital role in cancers and inducing ferroptosis in tumor cells has become a promising therapeutic method of cancer treatment. 42 A growing body of evidence indicates that ferroptosis is intricately linked to various biological processes in LUAD. 43 In this study, we revealed that RNF216 was involved in the regulation of ferroptosis in LUAD cells. Our results showed that RNF216 knockdown significantly promoted LUAD cell ferroptosis, as evidenced by elevated intracellular ROS, LPO, and Fe2+ levels, increased MDA content, and decreased GSH/GSSG ratio. In addition, the ferroptosis inhibitor, Fer-1, reversed the effects of RNF216 silencing on LUAD cell viability, migration, and invasion, indicating RNF216 might facilitate malignant phenotypes of LUAD cells by inhibiting LUAD cell ferroptosis. In vivo, RNF216 silencing also inhibited tumor growth and promoted ferroptosis.
The tumor suppressor, p53, plays an important role in cell processes. 44 Studies have shown that the regulation of p53 protein stability is crucial in the control of p53 activity. 45 p53 stability is critical in human malignancies, and its regulatory network is complex, with multiple genes involved in p53 ubiquitination degradation and stability. 46 Our study confirmed that RNF216 interacted with p53 in LUAD cells. In addition, RNF216 silencing increased p53 protein expression in LUAD cells and inhibited p53 ubiquitination and degradation in LUAD cells, consistent with a prior study that has identified that RNF216 serves as a moderator to target p53 ubiquitination and degradation in glioblastoma. 23
Increasing evidence showed that p53 can control cell survival, cell cycle, and cell apoptosis, and loss of p53 function has been demonstrated in various human cancers. 47 Besides, p53 can also modulate ferroptosis through transcriptional or posttranslational mechanisms. 48 Recently, studies have shown that p53 ubiquitination is negatively correlative to cell ferroptosis in some cancers, including breast cancer, 12 and cholangiocarcinoma. 11 However, p53 can also inhibit ferroptosis by blocking DPP4 activity. 49 The dual role of p53 makes it difficult to understand the exact molecular mechanisms controlling cell ferroptosis in LUAD. In our study, the p53 inhibitor, PFT-α, significantly reversed the effects of RNF216 silencing on LUAD cell viability, migration, invasion, and ferroptosis.
Therapeutically, our findings position RNF216 as a promising target for LUAD treatment. While current standard treatments for LUAD, including chemotherapy, mutation-dependent targeted therapies, and immunotherapy, have achieved measurable clinical success their long-term efficacy remains substantially limited by intrinsic and acquired resistance mechanisms. 50 Ferroptosis-based therapies hold great potential for overcoming resistance to these standard treatments. 51 Notably, RNF216-targeted therapy may offer synergistic advantages over conventional ferroptosis-inducing approaches by sensitizing tumor cells to ferroptosis while reactivating p53, a master regulator of multiple tumor-suppressive pathways. 52 Future research should prioritize developing combination strategies that integrate RNF216 inhibition with existing therapies in LUAD management.
Conclusion
This study highlights the oncogenic role of RNF216 in LUAD progression. Mechanically, RNF216 promotes p53 ubiquitination and degradation, thereby destabilizing p53 and facilitating LUAD cell proliferation, migration, and invasion by suppressing ferroptosis. By elucidating the direct influence of RNF216 on ferroptosis regulation via the p53 pathway, our findings provide critical insights into the molecular mechanisms driving LUAD progression. These insights lay the groundwork for the development of targeted therapeutic strategies, such as RNF216 inhibitors or p53-stabilizing agents, aimed at reactivating ferroptosis in LUAD cells. This approach holds promise as a novel avenue for ferroptosis-inducing therapies in LUAD treatment.
Footnotes
Statements and declarations
Author contributions
JW and WM designed this study. HW performed all the experiments and analyzed the data and prepared the figures. JJ drafted the initial manuscript. JZ reviewed and revised the manuscript. All authors read and approved the final manuscript.
Consent to publish
The authors affirm that all individual participants provided informed consent for publication.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by 2021PY029.
Conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All patient data were anonymized and maintained with confidentiality. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.