
Abstract
Doxorubicin (Dox) was reported to cause mitochondrial dysfunction and oxidative stress in cardiomyocytes, leading to cardiomyocyte apoptosis and ultimately heart failure. Serum and glucocorticoid inducible kinase 1 (SGK1) participates in the progression of various cardiovascular diseases. Thus, we aimed to explore the role and regulatory mechanism of SGK1 in Dox-induced cardiomyocyte injury. The expression of SGK1 was evaluated in blood samples of heart failure children, and in myocardial tissues and blood samples of Dox-induced rats. Subsequently, we treated cardiomyocytes with Dox in vitro. A gain-of-function assay was performed to assess the effects of SGK1 on mitochondrial dysfunction and oxidative stress in Dox-induced cardiomyocytes. Furthermore, the modulation of SGK1 on Neural precursor cell-expressed developmentally down-regulated 4 type 2 (NEDD4-2) expression and the subsequent Hippo pathway was validated. In our study, we found that SGK1 was downregulated in blood samples of heart failure children, as well as myocardial tissues and blood samples of Dox-induced rats. SGK1 overexpression alleviated the decreases of mitochondrial complex activity, mitochondrial membrane potential, adenosine triphosphate (ATP) content and ATP synthetase activity stimulated by Dox. Besides, SGK1 overexpression reversed the promoting effects of Dox on oxidative stress and apoptosis. Mechanistically, SGK1 overexpression inhibited the expression of NEDD4-2 and blocked the subsequent activation of Hippo pathway. NEDD4-2 overexpression or activation of Hippo reversed the protective effects of SGK1 overexpression on Dox-induced cardiomyocyte injury. In conclusion, our results revealed that SGK1 modulated mitochondrial dysfunction and oxidative stress in Dox-induced cardiomyocytes by regulating Hippo pathway via NEDD4-2.
Introduction
Heart failure is a common disease in the field of pediatric cardiology. 1 Therefore, it is essential to explore the molecular regulatory mechanism of heart failure. Doxorubicin (Dox), an effective antineoplastic antibiotic, induces cardiotoxicity and often results in degenerative cardiomyopathy, eventually leads to heart failure.2,3 Dox-induced rats and cardiomyocytes were often used as in vivo and in vitro heart failure models in many researches, respectively.4,5 Treatment with Dox triggered mitochondrial dysfunction, oxidative stress and apoptosis of cardiomyocytes.6,7 Cardiomyocyte apoptosis results in the attenuation of myocardial contractility, which plays a significant role in the pathogenesis of heart failure. 8 However, the specific molecular and regulatory mechanisms involved in Dox-induced heart failure remain to be further explored.
Mitochondria plays an essential part in heart metabolism and bioenergetics. 9 The imbalance of mitochondrial function is correlated with the loss of mitochondrial membrane potential (MMP), abnormal mitochondrial respiratory chain complex activity and inhibition of adenosine triphosphate (ATP) synthase activity. 10 Mitochondrial dysfunction is a critical event leading to Dox-induced cardiac injury. 11 Evidence from both in vivo and in vitro studies suggests that Dox causes mitochondrial dysfunction by inhibiting mitochondrial fusion and promoting mitochondrial fission. 12 Mitochondrial dysfunction results in metabolic disturbance and oxidative stress, impairs Ca2+ homeostasis and inflammation, so as to induce the development of heart failure. 13 Additionally, the oxidative stress of cardiomyocytes is also related to the pathological development of heart failure. 14 In heart failure patients, oxidative stress is triggered by reactive oxygen species (ROS) production in the myocardium, accompanying with cardiomyocyte apoptosis. 15
Serum and glucocorticoid inducible kinase 1 (SGK1), a member of the serine/threonine kinase gene family, is highly expressed in the heart 16 and is involved in various cellular processes. 17 SGK1 was reported to regulate mitochondrial function in renal diseases. 18 Besides, SGK1 is a stress response gene and closely related to the occurrence and development of many cardiac diseases. Studies have shown that SGK1 was linked to the increase of cardiac inflammation and fibrosis, which led to cardiac hypertrophy. 19 Moreover, SGK1 was involved in regulating inflammation and cell death in the ischemic-reperfused heart,20,21 and mediated the anti-autophagy effect of H2S in hypoxia/reoxygenation cardiomyocytes. 22 Importantly, SGK1 functioned importantly in cardiac arrhythmia treatment and activation of cardiac contractility.23,24 However, the role and potential regulatory mechanism of SGK1 in heart failure in children remain vague.
In our study, the expression of SGK1 was detected and the effects of SGK1 on mitochondrial dysfunction and oxidative stress in Dox-induced cardiomyocytes injury were validated. Moreover, we explored the potential regulatory mechanism of SGK1 in Dox-induced cardiomyocyte injury. We aimed to explore the molecular regulatory mechanism of Dox-induced cardiomyocyte injury and provide a theoretical basis for the development and therapy of heart failure in children.
Materials and Methods
Blood samples and collection
The blood samples were collected from 30 heart failure children (10 males and 20 females, aged from 3-16 years) and 30 healthy children (16 males and 14 females, aged from 2-16 years) in Xi’an Children’s Hospital, separately. Blood samples were centrifuged at 1000×g for 10 min, then the supernatant was collected and stored at −80°C. All heart failure cases were clinically diagnosed in strict accordance with the National Institute for Health and Clinical Excellence. The healthy children have no related cardiovascular diseases. Written informed consent was acquired from each patient. Our study was approved by Xi’an Children’s Hospital. All procedures conformed to the Declaration of Helsinki.
Animals and treatment
The rat model of heart failure was established according to a previous method. 4 Briefly, twenty 2- to 3-week-old Sprague-Dawley (SD) rats were purchased from Shanghai SLAC Laboratory Animal Co. Ltd (China). After adaptive feeding for 2–3 days prior to experiments, rats were randomly divided into two groups (n = 10). Group 1 (Negative control, NC) rats were treated with an equivalent volume of sterile saline solution; Group 2 (Dox-induced group, Dox) rats were intraperitoneally injected with Dox 2 times each week, with each injection of 2.5 mg/kg Dox. All rats were housed under a 12-h light/dark cycle (light on 8:00 am) in a temperature- and humidity-regulated cage and allowed food and water ad libitum. After treatment for three weeks, all rats were sacrificed under anesthesia with 2% isoflurane to relieve pain. Blood samples and myocardial tissues were collected and stored. All experiments conformed to the guidelines of Animal Research: Reporting of In Vivo Experiments (ARRIVE) and approved by Xi’an Children’s Hospital.
Cardiomyocyte isolation, treatment and transfection
Primary neonatal rat cardiomyocytes were isolated from 1- to 3-day-old SD rats according to previously reported. 25 Briefly, neonatal SD rats were disinfected with 75% ethanol and the hearts were quickly removed under sterile conditions. The isolated hearts were washed three times with PBS pre-cooled at 4°C to remove blood stains and cut into 1 mm3 pieces. Subsequently, these pieces were digested with 0.25% trypsin at 37°C for 10 min, then, 10% fetal bovine serum (FBS) was added into the supernatant to stop the digestion. The remaining tissue was digested with trypsin again at 37°C for 10 min and repeated several times until all tissues were digested. The cells were collected after centrifugation at 1000 r/min for 10 min, resuspended and enriched in Dulbecco modified Eagle medium (DMEM, Corning, Manassas, VA, USA) with 0.1 mM bromodeoxyuridine (Sigma‐Aldrich) at 37°C for 72 h.
Cardiomyocytes were treated with 0 μM, 0.5 μM, 1 μM, 2 μM of Dox for 24 h. The 2 μL synthesized pcDNA3.1-negative (NC), pcDNA3.1-SGK1 (pcSGK1), pcDNA3.1-NEDD4-2 (pcNEDD4-2) and pcDNA3.1-MST1 (pc-MST1) were transfected to cardiomyocytes using 1 μL Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific, USA) before Dox treatment for 24 h.
RT-qPCR
Total RNA was extracted from samples or cardiomyocytes using Trizol reagent (Life Technologies, USA). Then, the cDNA was generated from the RNA by using PrimeScript RT regent Kit (Takara Bio Inc, Japan). RT-qPCR was performed on a Bio-Rad CFX96 real-time PCR system (Bio-Rad, USA) by using SYBR Green (Takara Bio Inc, Japan). The mRNA expression was normalized to that of β-actin and calculated with the 2−ΔΔCt method.
Western blot assay
Total protein was extracted from cardiomyocytes and determined concentration using a BCA kit (Pierce, Shanghai, China). The equal amount of protein was separated by SDS-PAGE and transferred onto the PVDF membrane. After blocking, the membrane was incubated with primary antibodies at 4°C overnight, followed by incubation with HRP-conjugated secondary antibody. The immunoreactive blots were visualized by ECL plus Kit (Invitrogen, Thermo Fisher Scientific) and detected using a protein Imager (Tanon, Shanghai, China). β-actin was used as an internal control. The primary antibodies include SGK1 (1:500; Abcam, USA), NEDD4-2 (1:2000; Abcam, USA), ANP (1:1000; Abcam, USA), BNP (1:2000; Abcam, USA), β-MHC (1:1000; Abcam, USA), Acta-1 (1:1000; Abcam, USA), Bcl-2 (1:2000; Abcam, USA), Bax (1:1000; Abcam, USA), Caspase3 (1:2000; Abcam, USA), MST1 (1:1000; Abcam, USA), pMST1 (1:2000; Cell Signaling), LATS2 (1:1000; Abcam, USA), pLATS2 (1:2000; Cell Signaling), YAP1 (1:1000; Abcam, USA), pYAP1 (1:10000; Abcam, USA), β-actin(1:5000; Abcam, USA).
Cell viability assay
Cell viability was detected using a Cell Counting Kit‐8 (CCK‐8, Dojindo, Kumamoto, Japan) following the manufacturer’s instructions. Briefly, cardiomyocytes were grown in 96‐well plates (5000 cells per well) for 48 h. Then, 10 μL of CCK‐8 solution was added to each well, and the cells were further incubated for 2 h at 37°C. The absorbance of each well was measured at 450 nm using a microplate reader (Thermo Fisher Scientific, USA).
Cell apoptosis assay
Cell apoptosis was detected using Annexin V-FITC/PI Detection Kit (KeyGEN, China) in cardiomyocytes. Briefly, cells were exposed without or with Dox for 24 h and digested with EDTA-free trypsin (KeyGEN, China), and then washed three times with cold PBS. Subsequently, cells were resuspended at a density of 1 × 106 cells/mL in 500 μL binding buffer. After stained with Annexin V-FITC and propidium iodide (PI) for 20 min incubated in the dark at room temperature, cells were examined immediately by flow cytometry within 1 h.
Mitochondrial function detection
For Myocardial mitochondrial respiration complex, a MitoCheck Complex I Activity Assay Kit (Darmstadt, Germany) was used. For mitochondrial membrane potential (MMP), tetramethylrhodamine ethyl ester (perchlorate) (TMRE, Invitrogen, Thermo Fisher Scientific, USA) was used. An ATP assay kit (Beyotime, Shanghai, China) was used to measure the ATP content.
Oxidative stress measurement
The level of ROS was detected by using Reactive Oxygen Species Assay Kit (Beyotime, China). The content of catalase (CAT), superoxide dismutase (SOD) and malondialdehyde (MDA) were determined by using detection kits (Solarbio, Beijing, China) according to the manufacturer’s instructions.
Statistical analysis
All experiments were repeated at least three times. GraphPad Prism 8.2 (GraphPad Software, La Jolla, CA, USA) was used for statistical analysis. All data were expressed as mean ± SEM. Between groups comparisons and multiple groups were performed by either Student t-test and one-way ANOVA, respectively, with values of p < 0.05 considered as statistically significance.
Results
SGK1 was downregulated in tissue samples and Dox-induced cardiomyocytes
To explore the role of SGK1 in pediatric heart failure, blood samples of heart failure children were recruited. The results revealed that the mRNA expression level of SGK1 in blood samples of heart failure children was lower than healthy children (Figure 1(a)). Subsequently, we established a Dox-induced rat model as previous studies,4,5 and found that the expressions of SGK1 were also significantly downregulated in blood samples and myocardial tissues of Dox-induced rats. Moreover, we treated cardiomyocytes with Dox as a heart failure cell model. As shown in Figure 1(c), cell viability was dramatically decreased after Dox induction for 24 h in a dose-dependent manner. The mRNA and protein expressions of heart failure marker genes (ANP, BNP, β-MHC and Acta-1) were elevated by Dox, suggesting heart failure cell model was successfully established (Figure 1(d) and (e)). Interestingly, RT-qPCR and western blot assay results showed that both mRNA and protein expression levels of SGK1 were also declined in cardiomyocytes after stimulating with 1 μM or 2 μM Dox (Figure 1(f) and (g)). Cardiomyocytes were treated with 1 μM Dox for 24 h in subsequent experiments. SGK1 was downregulated in blood samples of heart failure children and Dox-induced cardiomyocytes. (a) Relative mRNA of SGK1 in blood samples from heart failure children and healthy children. ** stands for p < 0.01 vs health group. (b) Relative mRNA of SGK1 in myocardial tissues and blood samples of Dox-induced rats. ** stands for p < 0.01 vs NC group. (C) Cardiomyocyte viability after induction with 0 μM, 0.5 μM, 1 μM and 2 μM Dox for 24 h. ** stands for p < 0.01 vs 0 μM group. (D and E) The mRNA and protein expressions of heart failure marker genes (ANP, BNP, β-MHC and Acta-1) in cardiomyocytes with or without treatment 1 μM Dox for 24 h. ** stands for p < 0.01 vs control group. (F and G) The mRNA and protein expressions of SGK1 in cardiomyocytes stimulated with 0 μM, 0.5 μM, 1 μM, 2 μM Dox for 24 h. ** stands for p < 0.01 vs 0 μM group.
SGK1 overexpression alleviated oxidative stress and apoptosis of Dox-induced cardiomyocytes
To investigate the effects of SGK1 on Dox-induced cardiomyocytes, cardiomyocytes were transfected with NC or pcSGK1 before Dox treatment. As expected, the mRNA and protein expression levels of SGK1 were significantly increased in pcSGK1-transfected cells (Figure 2 A and B). Compared with control, the relative levels of ROS and MDA were elevated, while SOD and CAT levels were reduced by the treatment of Dox. However, these results were reversed by SGK1 upregulating (Figure 2(c)). Besides, treatment of Dox promoted the cardiomyocyte apoptosis accompanied with elevated BAX and cleaved-caspase 3 levels, as well as declined Bcl-2 level, which was rescued by SGK1 overexpression (Figure 2(d) and (e)). SGK1 overexpression alleviated oxidative stress and apoptosis of Dox-induced cardiomyocytes. (A and B) The mRNA and protein expressions of SGK1 in Dox-induced cardiomyocytes transfected with pcSGK1. ** stands for p < 0.01 vs Ctrl group. (c) Relative ROS, SOD, CAT and MDA levels in cardiomyocytes with or without SGK1 overexpression. (d) Cardiomyocyte apoptosis in cardiomyocytes with or without SGK1 overexpression. (f) Relative cell apoptosis related protein expressions in cardiomyocytes were detected by western blot assay. ** stands for p < 0.01 vs Ctrl group; ## stands for p < 0.01 vs Dox group.
SGK1 overexpression mitigated the mitochondrial dysfunction of Dox-induced cardiomyocytes
Previous studies suggested that mitochondrial dysfunction was associated with cell apoptosis.
5
We further explore the effect of SGK1 on mitochondrial function in Dox-induced cardiomyocytes. In our study, the activities of mitochondrial respiratory chain complex were inhibited by Dox treatment, which were reserved when cardiomyocytes were transfected with pcSGK1 before Dox treatment (Figure 3(a)). Furthermore, the treatment of Dox reduced ATP content, ATP synthetase and MMP, whereas the transfection of pcSGK1 abolished the inhibition of Dox exerted on ATP content, ATP synthetase and MMP (Figure 3(b)-(d)). SGK1 overexpression mitigated mitochondrial dysfunction of Dox-induced cardiomyocytes. (a) Relative mitochondrial complex activity in cardiomyocytes with or without SGK1 overexpression. (b) MMP in cardiomyocytes with or without SGK1 overexpression. (c) ATP content in cardiomyocytes with or without SGK1 overexpression. (d) Relative ATP synthetase level in cardiomyocytes with or without SGK1 overexpression. ** stands for p < 0.01 vs Ctrl group; ## stands for p < 0.01 vs Dox group.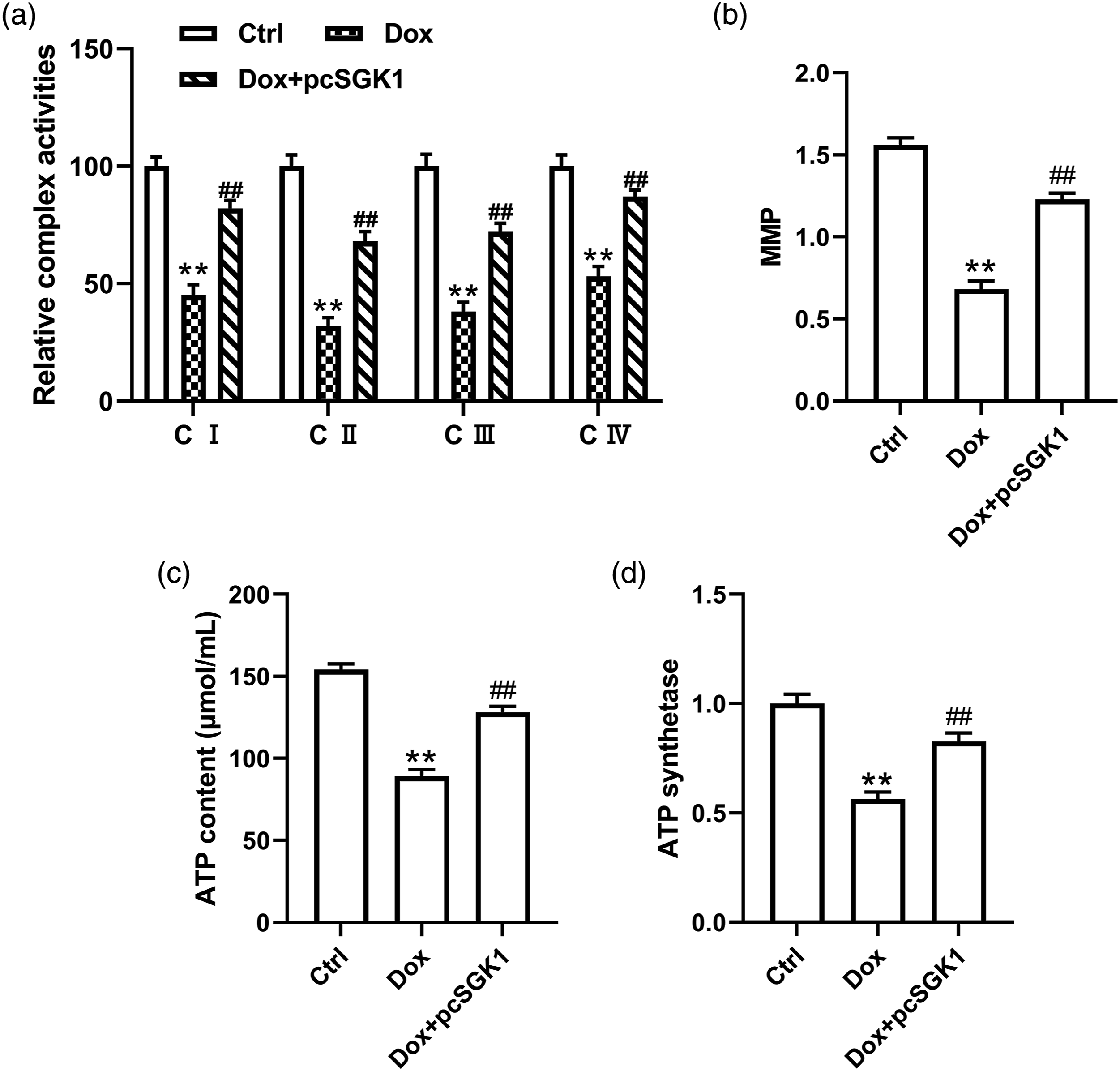
SGK1 regulated the Hippo signaling pathway via NEDD4-2
To explore the potential regulatory mechanism of SGK1 in heart failure, we detected the expression level of Neural precursor cell-expressed developmentally down-regulated 4 type 2 (NEDD4-2) in Dox-induced heart failure models. Previous study emphasized that SGK1 negatively regulated NEDD4-2 expression.
26
The results showed that the expression of NEDD4-2 was increased after Dox treatment (Figure 4(a)). The increased mRNA and protein expressions of NEDD4-2 were rescued by transfection with pcSGK1 (Figure 4(b) and (c)). Besides, NEDD4 regulated H2O2-induced cell proliferation and apoptosis via mediating Hippo pathway.
27
MST1 and LATS2 were the crucial components of the Hippo pathway and YAP1 was a main downstream effector of the Hippo pathway.
28
Western blot results showed that the phosphorylation levels of MST1, LATS2, and YAP1 was dramatically increased and the level of YAP1 was decreased by Dox induction. Subsequently, we boosted NEDD4-2 expression by transfecting with pcNEDD4-2. As we expected, the expression of NEDD4-2 was significantly upregulated in Dox-induced cells (Figure 4(d) and (e)). The Hippo pathway was partially inhibited by pcSGK1 pretreatment, which was reversed after transfection with pcNEDD4-2 (Figure 4(f)). SGK1 regulated Hippo signaling pathway via NEDD4-2. (a) The mRNA expression of NEDD4-2 in myocardial tissues of Dox-induced rats. ** stands for p < 0.01 vs Ctrl group. (b and c) The mRNA and protein expressions of NEDD4-2 in cardiomyocytes transfected with or without pcSGK1 before Dox treatment. ** stands for p < 0.01 vs Ctrl group; ## stands for p < 0.01 vs Dox group. (d and e) The mRNA and protein expressions of NEDD4-2 in Dox-treated cardiomyocytes transfected with or without pcNEDD4-2. ** stands for p < 0.01 vs Ctrl group. (f) The relative protein expressions of crucial components of Hippo pathway. ** stands for p < 0.01 vs Ctrl group; ## stands for p < 0.01 vs pcSGK1 group.
SGK1 regulated mitochondrial dysfunction and oxidative stress in Dox-induced cardiomyocytes by regulating the Hippo pathway via NEDD4-2
We further verified the regulatory mechanism of SGK1 in Dox-induced cardiomyocytes. We found that SGK1 overexpression inhibited the mRNA and protein expressions of heart failure marker genes after Dox treatment, which were rescued after transfection with pcNEDD4-2 or pcMST1 (Figure 5(a) and (b)). Besides, cell viability induced by pcSGK1 was partially inhibited by transfection with pcNEDD4-2 or pcMST1 (Figure 5(c)). Importantly, SGK1 overexpression reversed Dox-induced oxidative damage in cardiomyocytes, including mitochondrial dysfunction and oxidative stress. These effects were recused by transfection with pcNEDD4-2 or pcMST1 (Figure 5(d)-(h)). SGK1 regulated mitochondrial dysfunction and oxidative stress in Dox-induced cardiomyocytes by regulating Hippo pathway via NEDD4-2. (a and b) The mRNA and protein expressions of heart failure marker genes (ANP, BNP, β-MHC and Acta-1) in cardiomyocytes after treatment with Dox. (c) Cardiomyocyte viability after induction with Dox. (d) Relative mitochondrial complex activity in cardiomyocytes after Dox induction. (e) The MMP in cardiomyocytes after Dox stimulating. (f) The ATP content in cardiomyocytes with treatment of Dox. (f) Relative ROS, SOD, CAT and MDA levels in cardiomyocytes after treatment with Dox. (g) Cardiomyocyte apoptosis in cardiomyocytes with Dox treatment. (f) Relative cell apoptosis related protein expression in cardiomyocytes with stimulating by Dox. ** stands for p < 0.01 vs control; ## stands for p < 0.01 vs pcSGK1 group.
Discussion
SGK1 has been reportedly implicated in various cardiovascular diseases. 16 Evidence proved that SGK1 was decreased in diabetic cardiomyopathy and regulated cardiomyocyte survival.16,29 An early study showed that activation of SGK1 exerted protective effects on the myocardial infarction model by inhibiting cell apoptosis in vitro. 30 And knockdown of SGK1 aggravated cardiomyocyte damage in H/R injury. 21 These previous studies suggest that the decrease of SGK1 may be a mechanism resulting in heart failure. In the present study, we established Dox-induced heart failure models in vivo and in vitro. Dox-induced cardiomyocytes accompanied with increase of heart failure marker genes, inhibition of cell viability, mitochondrial function and oxidative stress, suggesting that Dox-induced cardiomyocytes as a heart failure cell model is feasible. Interestingly, based on these models, we validated that the expression of SGK1 was downregulated in heart failure children and in vivo models.
A growing body of evidence pointed out that inhibition of mitochondrial dysfunction and oxidative stress may be a way to alleviate heart disease. Huo and colleagues demonstrated that raloxifene alleviated pressure overload-induced heart failure by regulating mitochondrial dysfunction and oxidative stress. 14 Another study has shown that the knockdown of LncRNA KCNQ1OT1 inhibited cell apoptosis and increased MMP in a doxorubicin-induced heart failure cell model. 5 The current study confirmed that overexpression of SGK1 reduced mitochondrial dysfunction and oxidative stress in Dox-induced cardiomyocytes. SGK1 was a crucial regulator of mitochondrial dysfunction and oxidative stress. Jiang et al. demonstrated that SGK1 was essential for the induction of mitochondrial biogenesis and regulated renal tubular epithelial cell injury by mediating mitochondrial function. 18 The inhibition of SGK1 exacerbates mitochondrial dysfunction in the ischemic-reperfused heart. 20 Additionally, SGK1 inhibited oxidative stress in lung alveolar epithelial cells. 31 These findings support our conclusion that SGK1 mitigated Dox-induced cardiomyocyte injury by regulating mitochondrial dysfunction and oxidative stress.
Previous studies indicated that SGK1 inhibited the expression of NEDD4-2, 32 which was consistent with our finding. NEDD4-2 is an E3 ubiquitin ligase and regulates many cardiovascular diseases. 33 Luo et al. found that NEDD4-2 was increased and mediated the degradation of cardiac sodium channel in heart failure. 34 The inhibition of NEDD4 reduced cardiomyocyte autophagy by angiotensin Ⅱ stimulating 35 and exerted anti-apoptosis and protective effects on cardiomyocytes. 36 Consistently, we revealed that the enhanced expression of NEDD4-2 was discovered in Dox-induced cardiomyocytes and decreased by SGK1 overexpression, leading to mitochondrial dysfunction and oxidative stress. Notably, NEDD4-2 activated the Hippo pathway and mediated the regulation of SGK1 on mitochondrial dysfunction and oxidative stress in Dox-induced cardiomyocytes. Increasing evidence has indicated that the Hippo pathway plays a vital role in the development of heart failure. 37 MST1 (an upstream Hippo effector) reportedly induced cardiomyocyte apoptosis during the development of heart failure. 38 Subsequently, MST1 phosphorylation activated the phosphorylates of LATS1/2 and subsequent phosphorylates of YAP and TAZ, which also induced cardiomyocyte apoptosis. 39 The Hippo pathway was involved in the regulation of cell apoptosis and fibrosis in heart failure. 40 Moreover, Ikeda et al. suggested that excessive activation of Hippo was harmful to the heart and inhibition of Hippo was considered as a way to prevent the development of heart failure. 41 Additionally, Hippo has also been reported to be associated with mitochondrial dysfunction and oxidative stress. A recent report suggests that Emodin hindered oxidative stress via Hippo signaling pathway in the liver. 42 Inactivation of YAP, induced by Hippo activation, promoted mitochondrial dysfunction, accompanying with the decline of mitochondrial potential and the increase of mitochondrial oxidative stress in lung cancer cells. 43 The evidence strongly supports our findings.
Overall, our study revealed that the expression of SGK1 was decreased in heart failure children and Dox-induced models. Overexpression SGK1 suppressed the disorder of mitochondrial function and abnormal oxidative stress by inhibiting Hippo pathway via NEDD4-2 in Dox-induced cardiomyocytes. This might provide a molecular basis for the therapy of pediatric heart failure.
Footnotes
Author Contributions
Conceiving the study (YA), laboratory analyses (ZYZ, TTZ and ZZ), and statistical evaluation (TTZ), Writing the manuscript (ZYZ). All authors read and approved the final paper.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
The experimental protocol was established according to the ethical guidelines of the Helsinki Declaration and was approved by the Xi’an Children’s Hospital. Written informed consent was obtained from individual or guardian participants. All experiments conformed to the guidelines of Animal Research: Reporting of In Vivo Experiments (ARRIVE).
Data availablity
The data used to support the findings of this study are available from the corresponding author upon request.