
Abstract
Background
Ketamine, a non-competitive N-methyl-D-aspartate receptor (NMDAR) antagonist, is widely applicable to anesthesia, analgesia, and sedation. However, the function and mechanisms of ketamine in the long-term learning and memory function of neonatal mice are unclear.
Objective
The present study aims to investigate whether long-term learning and memory function will be affected by multiple ketamine exposures in the early development period.
Methods
The mRNA and protein levels were measured by RT-qPCR and western blot, respectively. The Morris Water Maze test was performed to assess spatial learning and memory.
Results
We identified that neonatal exposure to ketamine downsized the positive neurons for microtubule-associated protein doublecortin (DCX) and Ki67 in hippocampal dentate gyrus at the juvenile and late adolescence stages. Double-labeling tests demonstrated that the counts of Iba1+ cells and Ki67+ cells were pronouncedly diminished with exposure to ketamine. Further, qPCR assays to screen the key factors predisposing the populations and maturation of microglia exhibited remarkable decline of CX3CR1 mRNA levels in ketamine group versus the control group. The close relation of microglia to synaptic plasticity was depicted by the significantly downregulated synaptic plasticity-related proteins NR2B and PSD-95 subsequent to multiple exposures to ketamine. Finally, we found that both the protein and mRNA levels of BDNF were markedly decreased in ketamine group versus the control group.
Conclusion
We found that multiple exposures to ketamine in neonatal mice lead to spatial learning and memory dysfunction. The alterations of microglial development and function are the possible mechanisms of long-term learning and memory impairment.
Introduction
General anesthesia is performed in approximately 3.5 million infants and young children per annum, when their nervous system is still in neurodevelopment and is more fragile and vulnerable.1,2 Ketamine, a non-competitive N-methyl-D-aspartate receptor (NMDAR) antagonist, 3 is widely applicable to anesthesia, analgesia, and sedation, especially in the developing period. However, emerging studies of rodents and primates have demonstrated the neurotoxicity of postnatal ketamine anesthesia to the developing brain, as evidenced by induction of neuronal degeneration and apoptosis, retardation of neurogenesis, and impairment of synaptic plasticity, all of which are associated with long-lasting behavioral and cognitive deficits in adulthood.4,5
Microglial cells, which are distinct from other brain cells, originate from primitive myeloid progenitors in the yolk sac.6,7 In mice, the first microglia progenitors gain entry into the brain anlage via the leptomeninges and lateral ventricles around embryonic age 9 (E9) and disperse across the brain in all directions at varying speeds with different rates of proliferation.8,9 Studies show that the development of microglia undergoes three distinct temporal stages, defined as early microglia (until embryonic day 14), pre-microglia (from embryonic day 14 to several postnatal weeks), and adult microglia (from a few postnatal weeks onward). 10 In the initial two postnatal weeks of murine development, the microglial cells multiply, followed by a gradual decline by 50% from the third week to the sixth week, with microglial density stabilized thereon. 11
The microglia are subject to multiple factors with respect to proliferation, maturation, and homeostasis (e.g. the PU.1 and Irf8-dependent pathways are crucial to microglial embryonic development), which predispose the distinction from macrophages, 12 whereas other essential signaling pathways like CSF-1R and CX3CR1 are paramount in the maintenance of microglial physiology and microglia neuron interaction across the lifespan.13,14 Mice with CSF-1R knockout exhibited dramatically decreased microglia and impaired brain architecture. 15 Moreover, the CX3CL1/CX3CR1 pathway is also reportedly implicated in maintaining microglia at a resting state. 16 Mice deprived of CX3CR1 present with a variety of neuronal deficits, such as increased neuroinflammation, diminished synaptic pruning ability, and impaired neurogenesis, which are attributed to microglial dysfunctionality.17,18 Furthermore, the establishment of this microglia-specific gene expression profile in mice is restricted to the initial postnatal weeks, involving distinct microglial genes such as Olfml3, Tmem119, and Hexb,19,20 all serving as indicators of microglial maturation.
As a region closely related to learning and memory function, the subgranular zone (SGZ) of hippocampal dentate gyrus is among the regions most focused with respect to neurogenesis.21,22 Substantial hippocampal neurogenesis occurs during the first 2 weeks of life and approximately 70% of granular cells are generated in subgranular zone of the hippocampus, 23 some of which can migrate to the granular cell layer, and finally integrate into the hippocampal neural circuit, thereby establishing normal architecture of brain and the spatial learning and memory function. 24 During this critical phase, the brain possesses a high degree of plasticity, and microglia can affect the maturation of synaptic circuits via finely tuned mechanisms: their phagocytic activity allows for the elimination of inappropriate synapses and also affect the functional expression of synaptic proteins via mediator release. 25
Microglia, the only resident immune cells in the brain, is conventionally perceived as detrimental to neurodevelopment. 26 Multiple studies support that activated microglia release diverse inflammatory cytokines to create an environment that is inconducive to the survival of neonate neurons, or interferes with normal neurogenetic processes. 27 Additionally, aberrantly activated microglia excessively engulf normal neonate neurons, resulting in a downsized neuronal population. 28 However, recent studies have suggested that microglia exert a vital role in normal neurodevelopment and neuroplastic processes. 29 Microglia are reportedly associated with the severity of brain injury after cerebral ischemia, exacerbated with microglial scarcity. 30 In vitro, the population of cultured neural stem cells (NSCs) diminishes with the extended duration in contrast to the multiplication subsequent to addition of conditioned media of microglia-secreted factors, validating that microglia secrete certain factors to promote neurogenesis. 31 Further, emerging evidence indicates that microglia can secrete anti-inflammatory cytokines and neurotrophic factors, such as IGF-1, NGF, BDNF, etc.,32,33 with a pivotal role to play in neurodevelopment and neuroplastic processes.
Brain-derived neurotrophic factor (BDNF) serves a critical role in neurogenesis, learning-associated synaptic formation and is crucial for learning, memory, and advanced thinking. Albeit BDNF in brain appears to be chiefly neuronal in origin, it is still detectable in astrocytes and microglial cells. 34 Microglia, as a potential source of BDNF, were first discovered in culture and soon exhibited their indispensable neurotrophic effect in various neurological diseases, such as viral encephalitis, 35 traumatic injury, 36 ischemia, 37 multiple sclerosis, 38 Parkinson’s disease, 39 neuropathic pain, 40 etc. The CX3CR1−/− mice displayed reduced hippocampal neurogenesis, abnormal synaptic plasticity, and declined hippocampus-dependent learning tasks, compared with wild-type controls. Moreover, Cre-dependent depletion of BDNF from microglia results in hippocampal neurogenesis and synaptic formation abnormalities, and ultimately contributes to learning and memory dysfunction in adult mice, 41 which revealed potential functions of microglia in learning and memory via BDNF signaling. 42
However, prior researches on anesthetic-induced developmental neurotoxicity mainly focused on neuronal apoptosis, neuroinflammation, etc., with only a few experiments probing into the role of neurogenesis and synaptic plasticity, with microglia involved. Together, given the importance of microglia to brain development, plasticity and cognition, our study aimed to explore the effects of ketamine on the long-term learning and memory function of neonatal mice, as well as further explore the potential mechanisms associated with developmental and functional modification of microglia.
Materials and methods
Animals
All the animal experiments were approved by the Institutional Animal Care and Use Committee of Xuzhou Medical University. The Kunming murine pups, 6-day-old, were randomly allotted to the control or ketamine groups, which received intraperitoneal injection at an equal dose of normal saline or 80 mg/kg of ketamine diluted in 0.9% normal saline for five consecutive days,43–46 respectively, and were sacrificed by cervical dislocation following anesthesia at the following ages: postnatal day 11 (PND-11), PND-16, PND-21, PND-35, and PND-67. Throughout the anesthesia period, the pups were separated from the maternal mouse, with maintenance of the environmental temperature at 25 ± 1°C and meticulous surveillance of the skin color to avoid hypoxemia. Subsequent to emergence from anesthesia, the neonatal mice were group housed with the maternal mice until weaning, followed by routine feeding.
Morris water maze test
The MWM test was performed to assess spatial learning and memory. 47 The device comprised a circular black water tank of 100 cm in diameter, which was filled with water and maintained at a temperature of 20 ± 1°C. An escape platform (6 cm in diameter) was located 1.0 cm below (hidden) the water surface in a constant quadrant. The experiment lasted five consecutive days of training, with four trials per day. Mice were placed in the water with their nose pointing toward the wall at the starting point shifting between four constant locations at the pool rim, at an interval of 10 min for recovery. In case of failure to find the platform within 60 s, the animal would be guided gently to the platform and placed thereon for 15 s. On the sixth day, with the platform removed, each mouse was placed in the opposite quadrant to allow for free natation for 60 s to determine the search bias. Data were recorded with a computer connected to a video camera for collection of all variables of the MWM test. All behavioral experiments were conducted in a double-blind fashion and mice were tested by randomization.
Tissue preparation and immunofluorescence
The animals are subjected to deep anesthesia and transcardial perfusion with ice-cold 0.9% normal saline, followed by 4% paraformaldehyde in 0.1 M PBS (at pH 7.4). With mouse brains isolated and post-fixed overnight in fresh 4% paraformaldehyde, and thereafter transferred into 30% sucrose until sinkage. 48 The brain tissues were coronally sliced at the thickness of 40 μm on a microtome. Commencing from the initial section with visible ventral hippocampal commissure, the 6th-15th hippocampal slices were identified and stored in 0.01 M PBS solution. Subsequently, the target hippocampal sections (n = 4 mice per group; 4 sections per mice) were selected for blockage with 10% donkey serum in PBS with 0.4% Triton-X for 1 h at room temperature (r/t), followed by incubation with primary antibody against: Iba1 (24 h, dilution 1:400 at 4°C, ab5076, Abcam) to detect microglia, DCX (24 h, dilution 1:300 at 4°C, sc-8066, Santa Cruz) to detect neuroblasts and immature neurons, Ki67 (24 h, dilution 1:200 at 4°C, ab16667, Abcam) to detect proliferative cells, and BDNF (24 h, dilution 1:500 at 4°C, ab108319, Abcam). Having been rinsed thrice in PBS, sections were incubated with secondary fluorescent antibodies (Alexa546-labeled donkey anti-goat, Alexa488-labeled donkey anti-rabbit, or Alexa546-labeled donkey anti-mouse, dilution 1:200, Invitrogen) for 2 h at r/t. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI, KGA215-10, KeyGEN BioTECH), and the images were captured with a laser scanning confocal microscope (Fluoview 1000, Olympus). The positive cells in the hippocampal dentate gyrus were identified under a ×20 objective lens (620 μm x 620 μm) and manually counted using Image-Pro Plus software in a blinded manner.
Western blot
The animals were sacrificed for experiments at the following ages: postnatal day 11 (PND-11), PND-16, PND-21, PND-35, and PND-67, with bilateral hippocampal tissues meticulously isolated, followed by homogenization on ice with lysate buffer plus protease inhibitors. Following centrifugation at 12,000 r/min for 15 min at 4°C, the resultant supernatants were measured for protein concentrations with a BCA assay kit, with bovine serum albumin serving as a standard, and were transferred onto polyvinylidene fluoride membranes and blocked with blocking buffer for 1 h at r/t, followed by incubation with primary antibodies added to DCX (dilution 1:1000, sc-271390, Santa Cruz), Iba1 (dilution 1:500, ab5076, Abcam), BDNF (dilution 1:1000, ab108319, Abcam), CX3CR1 (dilution 1:800, ab8012, Abcam), PSD95 (dilution 1:1000, ab18258, Abcam), NMDA2B (dilution 1:1000, ab65783, Abcam), β-tubulin (dilution 1:1000, sc-5274, Santa), β-actin (dilution 1:2000, sc-47778, Santa Cruz), and GAPDH (dilution 1:2000, E12-052, Enogene) overnight at 4°C. Signals were detected with HRP-conjugated goat, rabbit or mouse antibodies followed by chemiluminescence with a New Super ECL kit (KeyGEN BioTECH). 49 Films were scanned and signals qualified by ImageJ (NIH, USA). Density of each band was normalized to controls.
Real-time PCR analysis
Mice were sacrificed at different time points: postnatal day 11 (PND-11), PND-16, PND-21, PND-35, and PND-67. Total RNAs were extracted from frozen hippocampal samples using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), then converted into cDNA by means of HiScript II Q RT SuperMix (R223-01, Vazyme) as per the manufacturer’s protocol. The cDNA were amplified with the primers as follows. CX3CR1 forward: 5′-GCA TTC TGA GCA GTT TCA CTC-3′, CX3CR1 reverse: 5′-ATC TGA TGC AAG AAC TCT GGG-3′; CSF1R forward: 5′-CGC CGA AGT GGG ATT CAA CG-3′, CSF1R reverse: 5′-CAG CGT TGA GAC TGA GAG CC-3′; TMEM119 forward: 5′-CTC CTG AAA GTC CCT GTG C-3′, TMEM119 reverse: 5′-CGT GGT ATC AAG GAG CAG TTA G-3′; OLFML3 forward: 5′-TTT TGT CAT GGA CGG GAC C-3′, OLFML3 reverse: 5′-CTA CTC TGA TCC CAT TGG-3′; HEXB forward: 5′-GAA GCT CCT GGT CTC CAT TAC-3′, HEXB reverse: 5′-GTC TCT AAA CCT CGT AAC GCT C-3′; BDNF forward: 5′-TAG CAA AAA GAG AAT TGG CTG-3′, BDNF reverse: 5′-TTT CAG GTC ATG GAT ATG TCC-3′; GAPDH forward: 5′-GGT GAA GGT CGG TGT GAA CG-3′, GAPDH reverse: 5′-CTC GCT CCT GGA AGA TGG TG-3′. The SYBR Color qPCR Master Mix was employed for all PCR reactions, which were analyzed with a LightCycler 480 (Roche). The PCR amplifications were performed at 95°C for 5 min, followed by 40 cycles at 95°C for 15 s, 60°C for 30 s, and 72°C for 60 s, respectively. On completion of qPCR, a melting curve of amplified products was determined. Data were collected with LightCycler 480 series software, and evaluated by the Comparative CT Method (2−ΔΔCT).
Statistical analysis
Statistical analysis was conducted with the aid of SPSS 16.0, and all graphs performed by GraphPad Prism 5.01. All data were tested for normality by the Kolmogorov-Smirnov test. Differences were evaluated with an Independent-Samples T test. A two-way ANOVA followed by Tukey’s post hoc test was adopted to analyze the difference in escape latency between the ketamine group and the control group in the MWM tests. Numerical data are presented as mean ± SD, and p < 0.05 is considered statistically significant.
Results
Neonatal ketamine exposure-induced impairment of spatial memory in mice at the juvenile and late adolescence stages
The effects of ketamine exposure on spatial learning and memory in mice were depicted in Figure 1 and the typical track charts were displayed in Figure 1(b). The latency to escape onto the hidden platform in both groups had a declining tendency with the training progression, indicative of successful training. During the 5 days of training, the latency to attain the hidden platform was significantly prolonged in ketamine group as compared with the control group, and the escape latency of juvenile mice in ketamine group was prolonged by 48.3% on the fifth day of training (Figure 1(c); ANOVA, Day 1: p > 0.05, Day 2–5: p < 0.05), indicating that neonatal ketamine exposure induced significant impairment of learning and memory functionalities during the juvenile stage. In the spatial probe test, as delineated in Figure 1(d) and (e), the number of target platform crossing and the duration spent in the quadrant zone within 60 s were significantly reduced in ketamine group versus the control group (Figure 1(d) and (e), two-sample t-test; p < 0.01 and p < 0.05), with similar results in the late adolescence stage (Figure 1(f) to (i)). Taken together, these data indicated that neonatal ketamine anesthesia could induce long-lasting impairment of spatial learning and memory in mice. Multiple exposure to ketamine anesthesia in neonatal mice induced spatial learning and memory impairment in the juvenile and late adolescence stages. (a) Flow chart of the experimental protocols. (b, f) Representative swimming paths of spatial exploration. (c, g) Ketamine anesthesia significantly prolonged the escape latency in MWM in the juvenile and late adolescence mice versus the control groups. (d, h) Numbers of crossing the previous platform within 60 s were significantly decreased in ketamine group. (e, i) The percentage of time spent in target quadrant was significantly reduced in ketamine group. Data were presented as mean ± SD (n = 20). Ctrl, Control group; KTM, Ketamine group. *p < 0.05, **p < 0.01 versus control group.
Ketamine anesthesia interfered with neurogenesis of hippocampal dentate gyrus
Immature neurons, within a few hours after their genesis, start to express the microtubule-associated protein doublecortin (DCX). The rate of hippocampal neurogenesis was determined from DCX-positive cells presented in the subgranular zone (SGZ) of the DG. As illustrated in Figure 2(b) and (c), immunofluorescence staining revealed that the count of DCX+ cells per SGZ was significantly diminished in ketamine group versus the control group, with the population of cells co-labeled for DCX+ and Ki67+ in PND-35 and PND-67 mice in ketamine group reduced by 46.8% and 42.4%, respectively (Figure 2(b) and (c); two-sample t-test, p < 0.05). Representative immunofluorescent images of coronal DG sections were exhibited in Figure 2(a). In addition, the expression of DCX protein normalized to the controls in the hippocampus of P35 and P67 mice in ketamine group was significantly downregulated (Figure 2(d); two-sample t-test; p < 0.05). These results disclosed that neonatal ketamine exposure inhibited the proliferation of immature neurons in hippocampal dentate gyrus at the juvenile and late adolescence stages. Effects of ketamine on the proliferation of immature neurons in the hippocampal dentate gyrus (DG). (a) Typical immunofluorescence of coronal DG sections from the PND-35 and PND-67 mice were stained for the immature neuron maker DCX (red) and the proliferation marker Ki67 (green). The solid arrows pointed to DCX+/Ki67+ dual-labeled cells and the scale bars were 100 μm. (b) Quantification of the DCX+ cells in DG revealed minor cell population in ketamine group versus the control group at PND-35 and PND-67 (n = 4). (c) Likewise, ketamine-exposed mice had reduced count of DCX+/Ki67+ cells in contrast with the controls (n = 4). (d) Brain tissues were isolated for western blot analysis, which revealed downregulation of protein level of DCX after ketamine exposures in mouse hippocampus at PND-35 and PND-67 (n = 8). Data were presented as mean ± SD. Ctrl, Control group; KTM, Ketamine group. *p < 0.05, **p < 0.01 versus control group.
Multiple exposures to ketamine in neonatal mice induced alteration of microglial development
Compared with the littermate controls, the ketamine-exposed mice exhibited pronounced reduction in the density of Iba1+ cells in at stages of P11, P16, P21, P35, and P67 (Figure 3(c); two-sample t-test, p < 0.05); whereas in the physiological state, the Iba1-labeled microglia in the hippocampal dentate gyrus from P16 mice were the most numerous. To determine whether multiple ketamine exposures could exert effects on the microglial proliferation, dual-labeling with antibodies against Ki67 and the microglial marker Iba1 were performed (Figure 3(a)), which revealed that the microglial proliferation in the dentate gyrus in the ketamine group was reduced by 43.5%, 39.5%, 29.4%, 41.7%, and 50.0%, respectively (Figure 3(b); two-sample t-test, p < 0.05). In parallel, compared with the controls, Iba1 expression in the hippocampus was also decreased in ketamine group at the afore-mentioned time points (Figure 3(d), two-sample t-test, p < 0.05). Persistent reduction in microglial population and retardation of microglial proliferation within the DG subregion in ketamine-exposed mice. (a) Representative images illustrated staining with the microglial marker Iba1 and proliferative marker Ki67 in mice at PND-11, PND-16, PND-21, PND-35, and PND-67, respectively. The solid arrows pointed to Iba1+/Ki67+ dual-labeled cells and the scale bars were 100 μm. (b and c) Quantification of both Iba1+ cells and Iba1+/Ki67+ cells revealed diminished cell counts in ketamine group versus the control group at different stages (n = 4). (d) The expression of Iba1 was downregulated after ketamine exposures (n = 8). The data were presented as the mean ± SD. Ctrl, Control group; KTM, Ketamine group. *p < 0.05, **p < 0.01 versus controls.
Multiple ketamine exposures induced reduction of the microglial CX3CR1 in mice
With respect to microglial development and maturation, diverse factors are involved, the key factors at different stages such as CX3CR1, CSF1R, TMEM119, OLFML3, and HEXB, which contributed to the microglial population and maturation, were assessed. The results of qPCR were shown in the Figure 4(a), revealing the decline of CX3CR1 mRNA levels in the hippocampus in ketamine group exclusively into the stage of PND-67 (Figure 4(a); two-sample t-test, p < 0.05). Moreover, western blot indicated that the expression of CX3CR1 protein in the bilateral hippocampus in mice at P11, P16, P21, P35 and P67 also decreased in the ketamine group compared to the control group (Figure 4(b); two-sample t-test, p < 0.05), which implied that CX3CR1 might exert a significance role in this process, thereby impairing microglial development. Early multiple ketamine anesthesia affected the expression of key factors of microglia. (a) qPCR revealed the decrease in mRNA levels of CX3CR1, CSF1R, TMEM119, OLFML3, HEXB in hippocampus of PND-11, PND-35, and PND-67 mice after ketamine exposures. (b) The protein levels of CX3CR1 were significantly decreased in ketamine group at PND-11, PND-16, PND-21, PND-35, and PND-67. Data were presented as mean ± SD (n = 8). Ctrl, Control group; KTM, Ketamine group. *p < 0.05, **p < 0.01 vs. control group.
Early multiple ketamine anesthesia modified the synaptic plasticity-related protein level in hippocampus
Western blot analyses were employed to detect the expression of NR2B and PSD-95 proteins in the hippocampus in both ketamine and control groups at the stages of PND-11, PND-16, PND-21, PND-35 and PND-67. As shown in Figure 5(a) and (b), the protein levels of NR2B and PSD-95 in the bilateral hippocampus were markedly downregulated in ketamine group versus the control at different time points, or rather, the expression of synaptic plasticity-related protein NR2B in the ketamine group was drastically decreased as of P11 till P35, with the most evident decline by 37.7% in P16 mice (Figure 5(a); two-sample t-test, p < 0.05). Likewise, the expression of PSD-95 in the ketamine group was also significantly decreased from P21 to P67, with the most decrease by 31.6% in P67 mice (Figure 5(b); two-sample t test, p < 0.05). Effects of multiple ketamine exposures on the expression of NR2B and PSD-95 proteins in the hippocampus. (a) The bilateral hippocampal expression levels of NR2B, measured by Western blotting at different time points, significantly decreased in P11, P16, P21 and P35 mice exposed to ketamine. (b) The expression levels of PSD-95 in the hippocampus were significantly decreased in P21, P35 and P67 ketamine-treated mice. Data were presented as mean ± SD (n = 8). Ctrl, Control group; KTM, Ketamine group. *p < 0.05, **p < 0.01 vs. control group.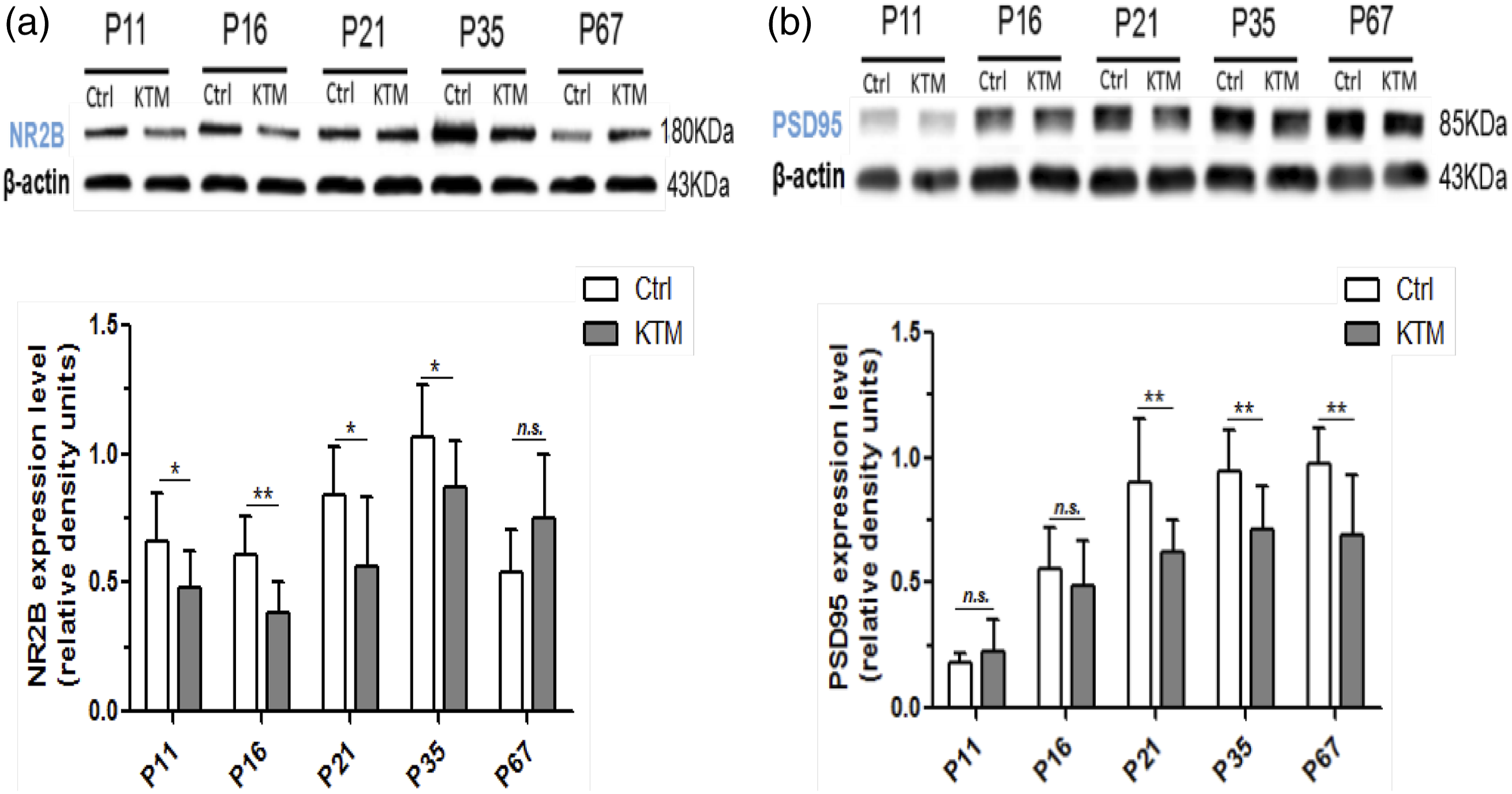
Multiple ketamine exposures contributed to physiological dysfunction of microglia
The above-mentioned results demonstrated that postnatal ketamine exposures induced the alteration of spatial learning and memory and hippocampal neurogenesis, along with the expression of synaptic plasticity-related protein NR2B and PSD-95. Nonetheless, the interactions between microglia and neuroplasticity remain masked. To address this mystery, we investigated the role of brain-derived neurotrophic factor (BDNF) therein. As presented in Figures 6(a) and (b), the BDNF protein and mRNA levels were markedly decreased in ketamine group versus the control group at PND-11, PND-16, PND-21, PND-35, PND-67, as measured by Western blot and q-PCR, respectively (Figure 6(a) and (b); two-sample t-test, p < 0.05), wherein the protein expression in the bilateral hippocampus decreased by 28.6% in the P16 mice in the ketamine group compared with the control group. The reduction was associated by a decrease in the population of Iba1+/BDNF+ cells in DG in ketamine mice, which implied the decline in BDNF level of microglia, as compared with controls (Figure 6(c); two-sample t-test, p < 0.05). Taken together, these findings confirmed the toxicity of postnatal ketamine anesthesia to microglial development and normal function, which might premise the impairment of neurogenesis and long-lasting cognitive deficits. Ketamine anesthesia-induced reduction of BDNF expression of microglia in mice. (a) The protein levels of BDNF were assessed by means of Western blot analysis at PND-11, PND-16, PND-21, PND-35, and PND-67 (n = 8). (b) qPCR analyses revealed the decline in the mRNA levels of BDNF in ketamine group versus the controls (n = 8). (c) The counts of Iba1+/BDNF+ cells at different stages were markedly reduced in ketamine group versus the control group (n = 4). (d) Representative images of Iba1 and BDNF expression were depicted. The arrows pointed to Iba1+/BDNF+ dual-labeled cells and the scale bars were 100 μm. Data were represented as mean ± SD. Ctrl, Control group; KTM, Ketamine group. *p < 0.05, **p < 0.01 versus controls.
Discussion
As a general anesthetic, ketamine is commonly applicable in clinical scenario. Nevertheless, postnatal ketamine anesthesia has been reported to exert neurotoxicity to the developing animal brain,50,51 with the mechanisms underpinning the neurogenesis and cognitive deficits in ketamine-exposed mice still remaining elusive. Microglia, conventionally deemed as detrimental, are nowadays acknowledged as the resident immune cells in the brain and essential to monitor the normal function of neurons and neuronal networks, as well as “shaping” of hippocampal neurogenesis. Due to the notional transition with respect to the role of microglia, 52 we proposed to explore the profiles of microglia in postnatal mice undergoing multiple ketamine anesthesia and the potential underlying mechanism herein.
As per literature retrieval, the half effective dose (ED50) of ketamine in anesthesia in mice, and meanwhile, given that both the peak of brain development and the proliferation and development of microglia are at approximately 2 weeks after birth, 45 we thereby selected the intraperitoneal dose of ketamine at 80 mg/kg.43,44 Moreover, based on our preliminary experiment, we designated intraperitoneal injection of ketamine at 80 mg/kg in the P6 mice to establish neonatal mouse models of long-term learning and memory dysfunction induced by multiple ketamine exposure. Our present study demonstrated that neonatal mice exposed to quintuple procedures of ketamine anesthesia at 80 mg/kg subsequently presented with spatial learning and memory impairment at the juvenile and late adolescence stages, with retardation of proliferation and reduction of immature neuronal population in the hippocampal dentate gyrus. A single intraperitoneal injection of propofol at the dose of 60 mg/kg reportedly decreased the number of BrdU-positive cells in the DG in neonatal mice 53 ; however, no significant alternation of BrdU-positive cells was identified in the DG between pups treated with propofol at 30 mg/kg or with identical volume of vehicle, suggesting that propofol suppressed the hippocampal neurogenesis in mice during an early postnatal stage in a dose-dependent manner. The in vitro evidence by Wu et al. 5 showed that the regulation of neurogenesis by ketamine was also time- and dose-dependent. In the rat model of schizophrenia, repeated application of subanesthetic doses of ketamine enhanced hippocampal neurogenesis. In contrast, Zhao et al. 51 described that ketamine administration in pregnant rats in the second trimester evoked long-lasting behavioral disorders in the offspring and decreased the number of DCX+/Brdu+ co-labeled cells in DG as well as the levels of BDNF, NR2B, and PSD95 in hippocampus, which is supportive of our experiments.
Of the factors in neuroplasticity, microglia, which were originally regarded as contributory to inflammation levels gained attention. 22 Monje et al. 54 reported that LPS-induced inflammation inhibited neurogenesis in the adult rat hippocampus via the microglial release of IL-6 and TNF-α. Additionally, microglia as the resident immune cells in brain impact the hippocampal neurogenesis via apoptosis-coupled phagocytosis. 55 In contrast to previous findings, microglia with a “resting” state also play diverse roles in neurogenesis and synaptic plasticity under normal physiological conditions, 22 as illustrated by microglia-affected expression of BDNF in enriched environment (EE), 56 which is related with learning, memory, and neuroplasticity. Additionally, recent studies demonstrate that microglia play an essential role in neurogenesis and synaptic plasticity, at least in part via microglial CX3CR1 signaling. 57 Meanwhile, BDNF is a critical mediator of neurogenesis, and Cre-dependent depletion of BDNF from microglia enhances the possibility that microglia have a potential function of shaping hippocampal neurogenesis via BDNF signaling. 42
To determine whether multiple ketamine exposure could exert any effect on microglia cells, we performed dual-labeling with antibodies against Ki67 and the microglial marker Iba1. The quantification of both Iba1+ cells and Iba1+/Ki67+ cells were decreased in ketamine group as compared with the control group at PND-11, PND-16, PND-21, PND-35, and PND-67, indicating the inhibition of microglia development. Moreover, the protein and mRNA levels of BDNF in ketamine group were significantly decreased versus the control group at PND-11, PND-16, PND-21, PND-35, PND-67, which were assessed by Western blot and qPCR, respectively. Notably, this decrease was associated with the reduction of the population of Iba1+/BDNF+ cells in ketamine mice as compared to controls, suggestive of the dysfunction of microglial cells.
Conclusion
The abovementioned findings authenticated that the alterations of microglial development and function might contribute to long-term learning and memory dysfunction in neonatal mice exposed to multiple ketamine manipulations. However, the small sample size limits the statistical power and future studies will be needed to confirm our results.
Supplemental Material
Supplemental Material - Implication of microglia in ketamine-induced long-term cognitive impairment in murine pups
Supplemental Material for Implication of microglia in ketamine-induced long-term cognitive impairment in murine pups by Y Yin, H Li, J Wang, Y Kong, J Chang and G Chu in Human & Experimental Toxicology
Footnotes
Authors’ contributions
YY, HL, JW and GC designed the present study. YK and JC performed the experiments and analyzed the data and prepared the figures. YY, HL and JW drafted the initial manuscript. GC reviewed and revised the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Science and technology development fund of Nanjing Medical University (NMUB2018068).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.