
Abstract
Background
As an aggressive human malignancy, esophageal squamous cell carcinoma (ESCC) is prevalent globally, especially in China. Verbascoside (VE) exerts anti-cancer effects in several human cancers. This work was to investigate the effects of VE on ESCC cells.
Methods
Esophageal squamous cell carcinoma cell proliferation, apoptosis, migration, and invasion were assessed by CCK-8, TUNEL, and Transwell assays. Gene and protein levels were detected by RT-qPCR and western blotting. CDC42 activity was evaluated by G-lisa assay.
Results
Verbascoside significantly inhibited cell proliferation, migration, and invasion and induced cell apoptosis in ESCC cells. Furthermore, it was found that VE markedly inhibited HMGB1 and RAGE expression in a dose-dependent manner. Besides, HMGB1/RAGE upregulation partially reversed the anti-cancer effects of VE on ESCC cells. VE repressed HMGB1/RAGE-induced CDC42 activation in ESCC cells. In addition, ML141-mediated CDC42 inactivation further enhanced the effect of VE on ESCC cell proliferation, apoptosis, migration, and invasion.
Conclusions
Our findings indicated that VE has significant anti-tumor potential in ESCC by suppressing HMGB1/RAGE-dependent CDC42 activation.
Introduction
Esophageal cancer (EC) is an aggressive digestive-tract malignancy with increasing incidence. 1 As the most prevalent histologic type of EC, esophageal squamous cell carcinoma (ESCC) accounts for about 80% of EC cases diagnosed globally. 2 ESCC manifests few symptoms at an early stage. 3 Hence, ESCC patients present with locally advanced tumors, metastasis, and unresectable tumors in most cases, with a 5-year survival rate below 25%. 4 Currently, the standard-of-care for patients with advanced ESCC is radiation and chemical therapies. 5 However, few drugs exert good long-term efficacy for ESCC therapy. 6 Therefore, it is imperative to find more therapeutic agents for ESCC treatment.
High mobility group box 1 (HMGB1), an abundant non-histone component of chromatin, 7 exists in almost all cell types and exerts intranuclear and extracellular functions in biological processes. 8 Emerging evidence indicates that co-expression of HMGB1 and receptor for advanced glycosylation end-products (RAGE, also known as AGER) is closely linked to cancer progression and poor outcomes of patients.9,10 Furthermore, obstruction of the HMGB1/RAGE signaling could diminish tumor growth and cancer progression in varied human cancers.11–13 Therefore, targeting specific signaling mediated by the HMGB1/RAGE complex might be a potential path to developing novel therapies for ESCC. Cell division cycle 42 (CDC42), a member of the Rho GTPase family, assumes a crucial role in tumorigenesis through regulating cytoskeletal dynamics. 14 Noteworthily, it was also identified that HMGB1-mediated RAGE activation may also induce CDC42 activation. 15 However, whether the HMGB1/RAGE/CDC42 signaling is implicated in ESCC progression needs to be determined.
Verbascoside, also known as Acteoside, is a phenylethanoid glycoside that can be found in several plant species. 16 Currently, VE has been demonstrated to exert cytotoxic and apoptotic effects in human cancers, including colorectal cancer, 17 oral squamous cell carcinoma, 18 breast cancer, 19 and glioblastoma. 20 Nevertheless, its effect and molecular mechanism in ESCC remain obscure. Since VE is a potent inhibitor for HMGB1, 21 we framed a hypothesis that VE could inhibit malignant behaviors of ESCC cells by suppressing HMGB1/RAGE expression and CDC42 activity.
This study was to explore whether VE exerts an antitumor effect on ESCC cells and identify an underlying molecular mechanism involved in VE-induced inhibition of the biological behaviors of ESCC cells. Our data showed that VE regulated ESCC cell proliferation, apoptosis, migration, and invasion by decreasing HMGB1/RAGE expression and inhibiting CDC42 activation, thus providing an experimental basis for VE as a promising drug for ESCC treatment and suggesting HMGB1/RAGE complex and CDC42 as potential therapeutic targets for ESCC.
Materials and methods
Cell culture and transfection
Esophageal squamous cell carcinoma cell lines (Eca-109 and TE-1) and normal esophageal epithelial cell line (Het-1A) were purchased from Cell Bank of the Chinese Academy of Science (Shanghai, China) and cultured in a humidified incubator (37°C; 5% CO2).
Empty pcDNA 3.1 vector (Vector) and pcDNA 3.1/HMGB1 overexpression vector (oe-HMGB1) were constructed by GenePharma (Shanghai, China). The above plasmids were transfected into ESCC cells via Lipofectamine 3000 (Invitrogen, USA) as per the manufacturer’s instructions.
Cell grouping and treatment
Eca-109 and TE-1 cells were divided into Control group, Vector group (transfected with Vector), oe-HMGB1 group (transfected with oe-HMGB1), oe-HMGB1+VE group (transfected with oe-HMGB1 and treated with VE at 50 μM), VE group (treated with VE), and VE+ML141 group (treated with VE at 50 μM and ML141 at 20 μ
RT-qPCR
Trizol reagent (Invitrogen) was adopted to extract total RNAs from cell lines. Next, cDNA was generated from total RNA via a cDNA synthesis kit (Takara). Afterwards, PCR was conducted on CFX Real-time PCR Detection System (Bio-Rad, USA) using Premix Ex Taq™ II (Takara).
Western blotting
Total proteins were extracted from cell lines. After concentration determination, total protein was separated via SDS-PAGE and transferred onto PVDF membranes. The membranes were washed twice with PBS, incubated with primary antibodies against HMGB1, RAGE and CDC42 overnight, and then incubated with secondary antibodies for 2 h. Finally, the membranes were analyzed with an enhanced chemiluminescence detection system (EMD Millipore, USA).
CCK-8 assay
To determine VE cytotoxicity in ESCC cells, cells were seeded into 96-well plates (5×103/well) and incubated with VE at different concentrations (0, 25, 50, and 100 μM) at 37°C for 24 h. Next, the CCK-8 reagent (10 μL/well) was supplemented. After incubation for another 2 h, absorbance at 450 nm was detected via a microplate reader (Thermo Plate, Germany).
To evaluate ESCC cell viability time-dependent study, cells were seeded into 96-well plates (5×103/well) and incubated with VE (50 μM) for 0, 24, 48, and 72 h. Subsequently, CCK-8 assays were performed as mentioned above.
TUNEL analysis
Cell apoptosis was determined by In Situ Cell Death Detection kit (Roche Diagnostics). Briefly, cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and then treated with TUNEL reaction mixture. TUNEL-positive cells were calculated under a fluorescence microscope.
Transwell assay
24-well transwell inserts (8 μm pore size; Corning) with or without Matrigel coating were used for cell invasion or migration assay. In brief, ESCC cells were resuspended in serum-free media and transferred to the upper chamber (2×104 cells). The lower chamber was supplemented with complete media containing 10% FBS. After incubation for 48 h, the invaded or migrated cells were fixed with 4% paraformaldehyde, stained with 0.1% crystal violet, and finally counted under a light microscope.
G-LISA assay
A CDC42-specific G-LISA Kit (Cytoskeleton, USA) was utilized to evaluate CDC42 activity as per standard instructions. Briefly, treated ESCC cells were rinsed twice with Locke’s solution and lysed in Cytoskeleton G-Lisa ® lysis buffer. Next, active GTP-bound CDC42 in the lysates was isolated and detected in an absorbance-based G-LISA system (Cytoskeleton) at 490 nm.
Statistical analysis
Each experiment was performed in triplicate. All data were expressed as Mean ± SD. Statistical analyses were conducted with SPSS 11.5. Differences between groups were analyzed by student’s t-test or one-way ANOVA. p-values less than 0.05 were deemed significant in statistics.
Results
VE inhibits malignant behaviors of ESCC cells
Deregulated proliferation and evasion of apoptosis are typical hallmarks of cancer cells.
22
To explore the effects of VE on ESCC cell proliferation and apoptosis, ECA-109 and TE-1 cells were subject to VE treatment at different concentrations (0–100 μM) for 24 h. As shown in Figure 1(a), V(E) exerted a suppressive effect on cell viability of both ECA-109 and TE-1 cell lines in a concentration-dependent manner. To verify the anti-proliferative role of VE, CCK-8 assay was also performed to assess the proliferative capability of ECA-109 and TE-1 cells treated with VE (50 μM). It was revealed that VE could inhibit ESCC cell proliferation in a time-dependent manner (Figure 1(b)). In addition, TUNEL assay also exhibited that VE could induce apoptosis in ECA-109 and TE-1 cells in a concentration-dependent manner (Figure 1(c) and (d)). Furthermore, Transwell assays disclosed that migrative and invasive potentials of ESCC cells were suppressed in VE-treated groups compared with the control group (Figure 1(e) and (f)). The above results indicated that VE exerted potential anti-tumor effects on ESCC cells. VE inhibits malignant behaviors of ESCC cells. (a) ECA-109 and TE-1 cells were subject to VE treatment (0, 25, 50, and 100 μM) for 24 h. Cell viability was detected by CCK-8. (b) ECA-109 and TE-1 cells were subject to VE treatment (50 μM) for 0, 24, 48, or 72 h. Cell viability was detected by CCK-8. (c and d) Cell apoptosis was detected by TUNEL. (e and f) Cell migration and invasion were assessed by Transwell assay. *p < .05; **p < .01; ***p < .001.
VE treatment reverses high HMGB1/RAGE expression in ESCC cells
As reported in previous studies, HMGB1 and RAGE exhibit elevated expression and oncogenic role in ESCC,23,24 implying the deep involvement of the HMGB1/RAGE axis in ESCC. Consistently, Western blotting and RT-qPCR assays showed that HMGB1 and RAGE levels were markedly elevated in ESCC cells (ECA-109 and TE-1) compared with normal cells (Het-1A) (Figure 2(a)). It has been identified that VE is an inhibitor of HMGB1/RAGE axis.
21
To examine the effects of VE administration on HMGB1 and RAGE expression in ESCC cells, two ESCC cell lines (ECA-109 and TE-1) were subject to VE treatment at different concentrations (0–100 μM). HMGB1 and RAGE levels were both declined in VE-treated ESCC cells in a dose-dependent manner (Figure 2(b) and (c)). Our results demonstrated that VE administration remarkably inhibited HMGB1/RAGE expression in ESCC cells. VE treatment reverses high HMGB1/RAGE expression in ESCC cells. (a and b) HMGB1 and RAGE protein levels in ESCC and normal cell lines by Western blotting and RT-qPCR. (b) HMGB1 and RAGE protein and mRNA levels in ECA-109 cells subject to VE treatment (0, 25, 50, and 100 μM). (c) HMGB1 and RAGE protein and mRNA levels in TE-1 cells subject to VE treatment (0, 25, 50, and 100 μM). *p < .05; **p < .01; ***p < .001.
HMGB1/RAGE upregulation abates VE-induced inhibitory effects on ESCC cell proliferation, migration and invasion
To figure out whether VE exerts its anticancer effect on ESCC cells via the HMGB1/RAGE axis, ESCC cells were transfected with Vector or oe-HMGB1 and then treated with VE (50 μM). Western blotting and RT-qPCR results exhibited that HMGB1 overexpression significantly upregulated HMGB1 and RAGE levels in ESCC cells (Figure 3(a)). As illustrated in Figure 3(b), HMGB1 and RAGE levels in ESCC cells were significantly increased after HMGB1 overexpression, which was partly reversed by VE treatment. Then, ECA-109 and TE-1 cells were divided into Control, VE, and oe-HMGB1+VE groups. Rescue assays further demonstrated that HMGB1 upregulation partially abolished the impact of VE treatment on ESCC cell proliferation, apoptosis, migration, and invasion (Figure 3(c) to (f)). To sum up, VE restrained malignant behaviors of ESCC cells by inhibiting the HMGB1/RAGE axis. HMGB1/RAGE upregulation abates VE-induced inhibitory effects on ESCC cell proliferation, migration and invasion. (a) HMGB1 and RAGE protein and mRNA levels in ECA-109 and TE-1 cells transfected with Vector or oe-HMGB1. (b) HMGB1 and RAGE protein and mRNA levels in ECA-109 and TE-1 cells in Vector, oe-HMGB1, or oe-HMGB1+VE groups. (c–f) Cell proliferation, apoptosis, migration, and invasion of ECA-109 and TE-1 cells in Control, VE, or oe-HMGB1+VE groups. *p < .05; **p < .01; ***p < .001.
VE inhibits HMGB1/RAGE-mediated CDC42 activation in ESCC cells
CDC42 is a downstream target of the HMGB1/RAGE axis and acts as an oncogene in ESCC.15,25 Therefore, we further explored whether VE could affect HMGB1/RAGE-induced CDC42 activation in ESCC cells. First, ECA-109 and TE-1 cells were subject to VE treatment (50 μM). It was found that VE treatment inhibited both CDC42 expression and activity in ESCC cells (Figure 4(a) and (b)), suggesting VE promoted CDC42 inactivation in ESCC. Next, ECA-109 and TE-1 cells were transfected with Vector or oe-HMGB1 and then treated with VE (50 μM). It was shown that HMGB1 upregulation significantly increased CDC42 levels and promoted CDC42 activation in ESCC cells, while VE treatment partially reversed such effects (Figure 4(c) and (d)). Taken together, our results indicated that VE inhibited CDC42 activation in ESCC cells by suppressing the HMGB1/RAGE axis. VE inhibits HMGB1/RAGE-mediated CDC42 activation in ESCC cells. (a) CDC42 protein and mRNA levels in ECA-109 and TE-1 cells. (b) CDC42 activation in ECA-109 and TE-1 cells was analyzed by G-LISA. (c) CDC42 protein and mRNA levels in ECA-109 and TE-1 cells in each group. (d) CDC42 activation in ECA-109 and TE-1 cells in each group. *p < .05; **p < .01; ***p < .001.
CDC42 inactivation enhances the anti-proliferative, anti-metastatic, and apoptotic effects of VE on ESCC cells
To verify the involvement of CDC42 activation in VE-induced suppression on biological behaviors of ESCC cells, a selective CDC42 inhibitor, ML141,
26
was used. Firstly, ECA-109 and TE-1 cells were treated with ML141. As shown in Figure 5(a) and (b), ML141 suppressed CDC42 level and activity in ECA-109 and TE-1 cells, affirming the inhibitory effect of ML141 on CDC42 activation. Then, ECA-109 and TE-1 cells were divided into Control, VE, and VE+ML141 groups. As illustrated in Figure 5(c) to (f), ML141 reinforced VE-induced inhibitory effects on cell proliferative, migration, invasion, and promoting effect on cell apoptosis. All these results indicated that ML141-mediated CDC42 deactivation further enhanced VE-induced anti-tumor effects on ESCC cells. CDC42 inactivation enhances the anti-proliferative, anti-metastatic, and apoptotic effects of VE on ESCC cells. (a) CDC42 protein and mRNA levels in ECA-109 and TE-1 cells treated with ML141. (b) CDC42 activation in ECA-109 and TE-1 cells. (c–f) Cell proliferation, apoptosis, migration, and invasion of ECA-109 and TE-1 cells in Control, VE, or VE+ML141 groups. *p < .05; **p < .01; ***p < .001.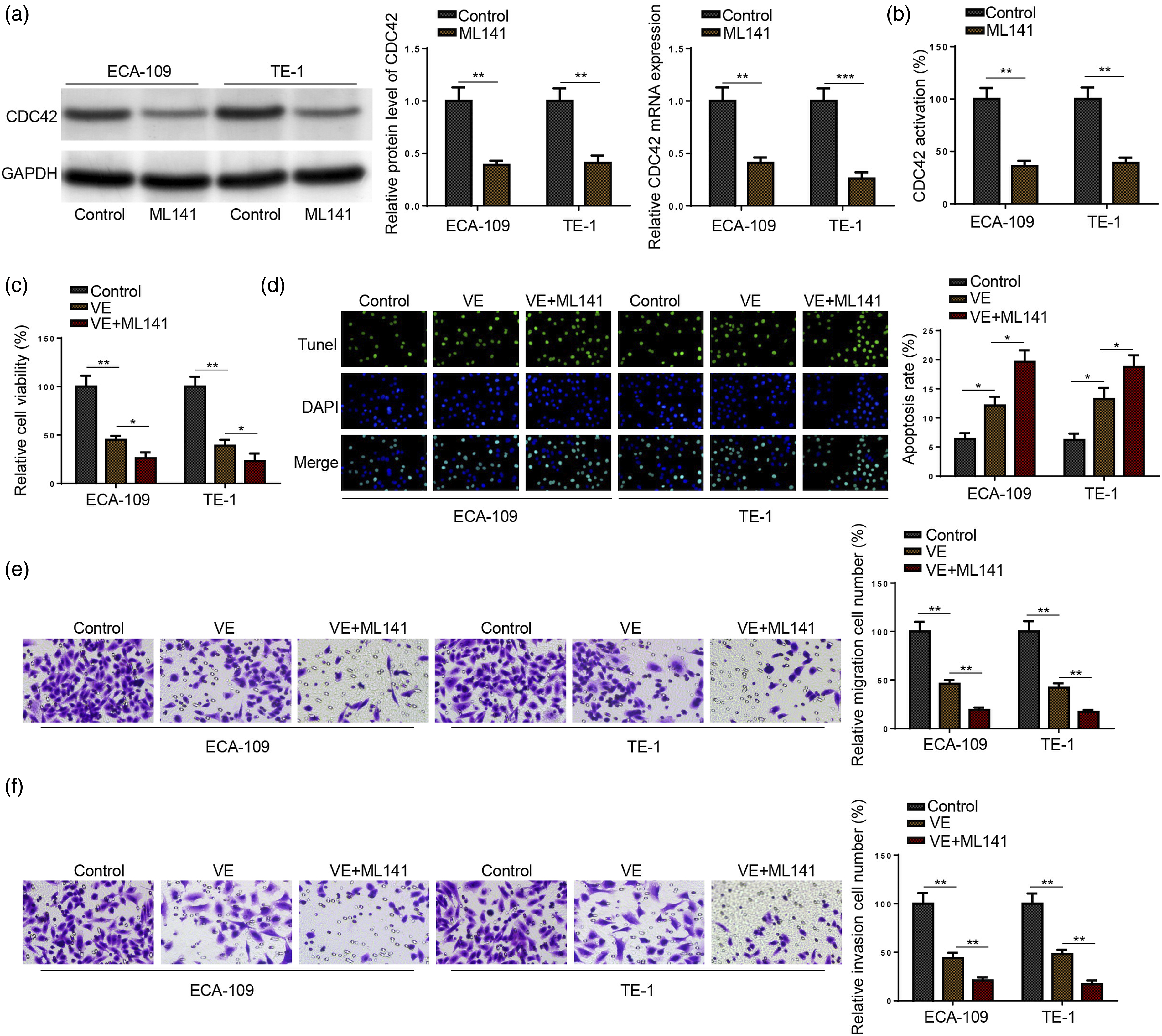
Discussion
As a fatal digestive-tract malignancy, ESCC endangers public health. 27 Although chemical therapy is for ESCC treatment, its clinical application is still limited due to chemoresistance and adverse effects. 28 Therefore, it is urgently needed to discover novel anti-cancer drugs for ESCC. It has been demonstrated that VE is a potential antitumor agent for several human cancers. For example, VE inhibits melanoma tumor growth by modulating the ERβ-Ras/Raf1-STAT3 signaling. 29 Besides, VE exerts an anti-tumor effect in human oral squamous cell carcinoma by inhibiting metastasis and inducing apoptosis. 16 In addition, VE also synergizes with temozolomide to exert anti-proliferative and apoptotic effects on glioblastoma cells. 30 In the present study, it was observed that VE administration could suppress cell growth, invasion, and migration and promote apoptosis in ESCC cells.
HMGB1/RAGE axis plays a crucial role in promoting tumor initiation and progression. 31 Previous studies have identified that HMGB1 and RAGE are both highly expressed in ESCC and contribute to malignant behaviors of ESCC cells,23,24 indicating HMGB1/RAGE axis could be a latent target for ESCC treatment. Furthermore, a study has identified that VE inhibits prostate cancer progression by regulating the HMGB1/RAGE/TGF-β signaling, 32 implying VE may exert anti-cancer effects in human cancers as an HMGB1/RAGE inhibitor. Therefore, we assumed that VE could mitigate malignant of ESCC cells via inhibiting HMGB1/RAGE. Consistent with previous studies, HMGB1 and RAGE levels were significantly higher in ESCC cells than in normal cells. Besides, we treated ESCC cells with VE at different concentrations and found VE induced a significant decline of HMGB1 and RAGE levels in ESCC cells in a dose-dependent manner. Moreover, rescue assays demonstrated that HMGB1 overexpression partially abated the impact of VE on ESCC cell proliferation, apoptosis, migration, and invasion. Therefore, VE might exert its antitumor effects on ESCC cells via interfering with the HMGB1/RAGE complex.
CDC42 is overexpressed and serves as an oncogene in diverse human malignancies. 33 Also, CDC42 deactivation may prevent tumor cell proliferation, thus enhancing the effectiveness of antitumor agents.34,35 To cite an instance, GINS4-induced CDC42 activation facilitates cell proliferation and metastasis in gastric cancer. 36 Besides, MBQ-167-mediated CDC42 inactivation could impair triple-negative breast cancer tumor growth and metastasis. 37 In addition, combined administration of dihydroartemisinin and resveratrol depresses cancer cell metastasis by targeting CDC42. 38 As one of the three prototypical Rho GTPases, CDC42 has attracted increasing interest as a potential target for the discovery of anti-cancer drugs. 39 Prior studies have identified CDC42 as a downstream target of the HMGB1/RAGE axis and an oncogene in ESCC.15,25 Hence, CDC42 may also be involved in VE-mediated anti-cancer effects in ESCC. Herein, VE significantly inhibited CDC42 expression and activity in ESCC cells. Additionally, HMGB1 overexpression substantially promoted CDC42 expression and activity in ESCC cells, whereas HMGB1/RAGE-induced CDC42 upregulation and activation was remarkably abated by VE treatment. Furthermore, functional assays further demonstrated that ML141-induced CDC42 inactivation could reinforce the impact of VE on ESCC cell proliferation, migration, invasion, and apoptosis. To sum up, our results showed that VE-induced anti-cancer effects on ESCC cells was mediated by inhibition of HMGB1/RAGE expression and CDC42 activity.
Conclusion
In the present study, we demonstrated that VE inhibited HMGB1/RAGE and CDC42 activation, consequently attenuating malignant behaviors of ESCC cells, suggesting VE treatment could be an efficient adjuvant therapy for ESCC. However, there is one key limitation in our study: all our results were obtained from in vitro experiments. In future studies, our findings will be further verified in vivo.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.