
Abstract
Introduction
Sepsis is an immunological response leading to multi-organ failure. 1 Pro- and anti-inflammatory responses result in a dysregulated reaction in sepsis. This reaction belongs to the host against the developing infection. 2 In the pathology of sepsis, the intracellular signaling process leads to expressional changes of genes contributing to adaptive immunity and inflammation. In the early stage of sepsis, the irregular inflammatory response may cause a low level of peripheral vasodilation associated with abnormal platelet stimulation.3,4 In the current literature, there are no specific FDA-approved therapeutic agents for the treatment of sepsis. Accordingly, the complications that develop in sepsis could be attenuated by therapeutic approaches that regulate immune responses, such as reducing the levels of pro-inflammatory mediators. 5
Sepsis initiates a complex immune response that changes over time through pro-inflammatory and anti-inflammatory mechanisms. As a result, septic patients quickly show signs of immunosuppression. 6 Immune suppression due to sepsis can lead to an uncontrolled inflammatory response. 7 The pathology of sepsis consists of an initial hyperinflammatory phase followed by a longer-lasting immunosuppressive phase. 8 Sepsis-induced immunosuppression may occur through mechanisms such as destruction of immune cells by apoptosis and increased expression of regulatory T (Treg) cells. 9 In this context, the stimulation of the complement system, coagulation system, vascular endothelium, neutrophils, and platelets may also play a role in the pro-inflammatory responses that occur in sepsis. 10
A disruption of the inflammatory balance is seen as the most critical characteristic in the pathogenesis of sepsis. A disruption of this order can be caused by pathogens such as bacteria, fungi, parasites, and viruses. 11 The first acute host response to these pathogens is reported as the engulfment of the pathogens by macrophages and production of a variety of pro-inflammatory cytokines. Cytokine storms can be triggered by the production of pro-inflammatory cytokines, or the innate immune system can be stimulated.12,13 Additionally, the constant stimulation of neutrophils and macrophages/monocytes can also lead to the acceleration of the septic response. Accordingly, the delayed apoptosis of neutrophils, upregulation of lymphocyte-associated costimulatory molecules, and increased necrosis of tissues seem to be associated with factors leading to the pathogenesis of sepsis. 14
In sepsis, the innate immune response that enables the invasion of pathogens is attributed to the development of the pathology.
15
Triggering the cellular pathway of the immune system causes an excessive release of cytokines, chemokines, and other inflammatory mediators such as prostaglandins. Cytokines can regulate inflammatory responses by transporting immune cells to the site of infection. Inflammatory response prevents infection by controlling the localized regions. Additionally, irregular cytokine release may lead to endothelial dysfunction associated with vasodilation and increased capillary permeability (Figure 1). Accordingly, the release of inflammatory mediators in the environment is related to hypotension, hemoconcentration, macromolecular extravasation, and edema in sepsis.16,17 In this review, we discuss inflammation mediators in sepsis with the current literature (Figure 2). Role of inflammatory mediators in the pathology of sepsis. (a) Immunologic changes in the body due to the inflammatory response in sepsis. Stimulation of TFs associated with an uncontrolled inflammatory response in sepsis may induce IL-6 expression. Cytokine production occurs in the region where the infection is localized. Diapedesis of monocytes is observed with stimulation of IL-6 expression. Additionally, ROS may cause changes in the microcirculation in sepsis through oxidative stress as a result of the inflammatory response. (b) The role of NK cells in regulating inflammatory mediator release in sepsis. During sepsis, NK cells can induce changes in IFN-γ and TNF-α, leading to cytokine release and microbial death. Here, the function of IL-2, IL-12, and IL-18 cytokines can be reduced by macrophages. Decreased immune function may affect complications associated with sepsis, resulting in long-term mortality (IL-2, Interleukin 2; IL-12, Interleukin 12; IL-18, Interleukin 18; IL-6, Interleukin 6; NK, Natural killer cells; TF, Tissue factor; ROS, Reactive oxygen species; IFN-γ, Interferon gamma; TNF-α, Tumor necrosis factor alpha). Role of inflammatory mediators in the pathology of sepsis. (a) Immunologic changes in the body due to the inflammatory response in sepsis. Stimulation of TFs associated with an uncontrolled inflammatory response in sepsis may induce IL-6 expression. Cytokine production occurs in the region where the infection is localized. Diapedesis of monocytes is observed with stimulation of IL-6 expression. Additionally, ROS may cause changes in the microcirculation in sepsis through oxidative stress as a result of the inflammatory response. (b) The role of NK cells in regulating inflammatory mediator release in sepsis. During sepsis, NK cells can induce changes in IFN-γ and TNF-α, leading to cytokine release and microbial death. Here, the function of IL-2, IL-12, and IL-18 cytokines can be reduced by macrophages. Decreased immune function may affect complications associated with sepsis, resulting in long-term mortality (IL-2, Interleukin 2; IL-12, Interleukin 12; IL-18, Interleukin 18; IL-6, Interleukin 6; NK, Natural killer cells; TF, Tissue factor; ROS, Reactive oxygen species; IFN-γ, Interferon gamma; TNF-α, Tumor necrosis factor alpha).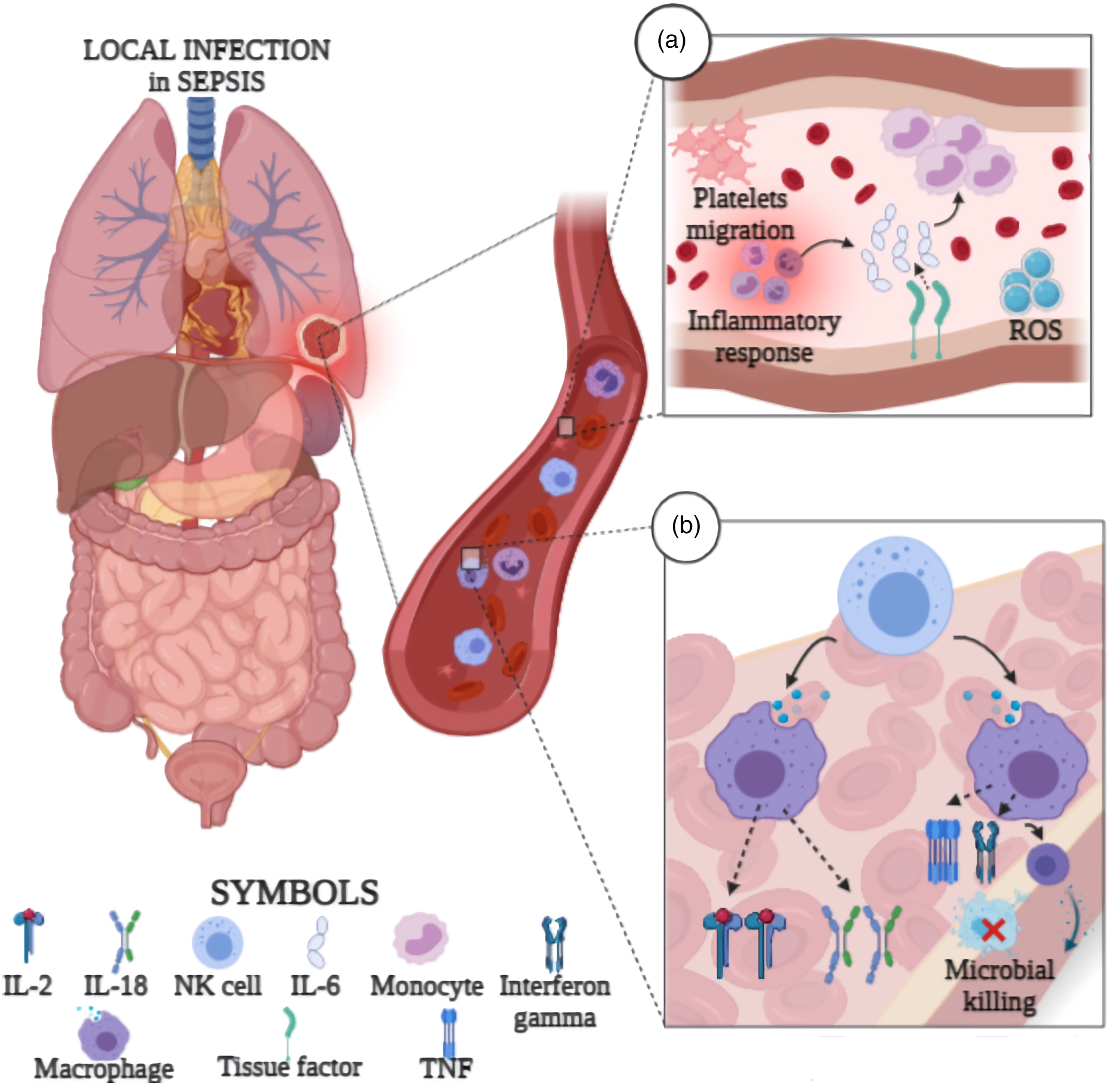
The role of cytokines in sepsis pathology
In the mechanism of sepsis, the pro-inflammatory response has a balance with certain anti-inflammatory cytokines such as interleukin 10 (IL-10), transforming growth factor-beta (TGF-β), and interleukin 4 (IL-4), restoring the immunological balance.
18
Sepsis lead
Data reported in animal studies have indicated that various processes such as inflammation, oxidative stress, and autophagy are involved in the mechanisms causing cardiac damage and dysfunction in sepsis.30–32 IL-16 expression from cardiac tissue and serum was observed to result in an increase in LPS-induced sepsis, while the increase in IL-16 expression was reduced by superoxide dismutase (SOD). IL-16 expression can be regulated by oxidative stress parameters in sepsis cases. Additionally, the nuclear factor erythroid 2 (Nrf2) pathways may be protectively mediated against sepsis-induced cardiac injury in anti-IL-16 therapy. 33 In a clinical study conducted with 32 sepsis patients, there was no significant difference in the expressions of antioxidant glutathione (GSH) and malondialdehyde (MDA) in peripheral blood. The absence of significant data about the relationships between oxidative stress markers, GSH, and MDA indicates the necessity of new studies on the effectiveness of oxidative stress in sepsis. Furthermore, sepsis-associated encephalopathy was alleviated by an anti-oxidant, ginsenoside, by suppressing oxidative stress. 34 Accordingly, oxidative stress may affect cardiac sepsis complications through inflammatory cytokines. Additionally, the Nrf2 pathway may be involved in the oxidative stress-related mechanism in cardiac damage in sepsis.
Studies show that inflammatory cytokines such as IL-10, IL-6, and TNF-α may be associated with the pathology of sepsis. The p65 pathway in the setting of sepsis with cardiac damage modulates M1 macrophage differentiation. 25 Another study showed that the pretreatment of IL-6 deficiency with a low dose of LPS in young mice reduced the protein kinase B (AKT)-related cell-protective effect. 26 These findings indicate the role of the AKT signaling pathway in cardiac dysfunction of cytokines such as IL-5 and IL-6 in sepsis. However, the role of M1 and M2 macrophage cells that regulate cytokine expression in inflammatory differentiation needs additional evidence to be reached with further studies. The potential correlation of oxidative stress and apoptosis molecules with inflammatory cytokines could be determined in cardiac damage. Hence, determining the complex factors regulating cytokines in sepsis may contribute to the improvement of the prognosis.
The role of chemokines in the pathology of sepsis
Peptide chemokines have a high concentration in circulation, and they suppress local inflammation by desensitization. 35 These peptides are secreted by tissue cells, leukocytes, and triggered epithelial cells. 36 Chemokines are defined in four distinct subfamilies such as CC chemokines, CXC chemokines, CX3C chemokines, and C chemokines due to the highly conserved presence of the first two cysteine residues. 37 Therefore, the chemokine family is critical in the pathology of sepsis.
The effectiveness of CC and CXC chemokine levels on disease severity was investigated in 58 children with meningococcal sepsis. Nine sepsis patients who died were reported to have significantly higher serum levels of CCL1, CCL2, CCL8, CCL20, and chemokines including CXCL8, CXCL10, and CXCL12 in comparison to surviving sepsis patients. Based on these results, chemokine levels in sepsis may be key candidates for prognostic practice based on laboratory parameters. 38 CXCR2 receptors are associated with the regulation of neutrophil uptake. 39 The efficacy of CXCR2 was investigated for differentiating sepsis from infection via neutrophil dysfunction. At disease onset, the CXCR2 level decreases with a dose–response relationship depending on the clinical course. Among the neutrophil functions examined in this study, there was a decrease in the CXCR2 surface level associated with sepsis. Accordingly, the decrease in the CXCR2 surface level is also associated with sepsis in the presence of infection. 40 On the other hand, some studies on changes in the surface levels of the CD64 and CCR2 chemokine families state that these changes are not associated with sepsis. In this context, the increase in CCR2 surface levels may be related to infection rather than sepsis, while the in CD64 surface levels may not. This conclusion is a dose–response relationship due to infection.40–42
Gain-of-function in CCR2 helps in the definition of the binding of polymorph nuclear leukocytes (PMNs). Blood PMNs and endothelial cells can put tissues at risk of injury due to an intense inflammatory response during sepsis induced by cecal ligation and puncture (CLP). 43 Based on these data, varying chemokine levels are stated to have a prognostic value in septic patients. The relationship of CCR2 in sepsis with PMN at varying surface levels needs to be evaluated in detail with future studies.
The effect of sepsis-associated encephalopathy (SAE) on hippocampal neurogenesis in the case of CXCR5 knockdown was investigated, and it was shown that reduced memory capacity and learning deficits are associated with CLP-induced sepsis. 32 CXCR5 may function in reducing the maintenance of immature neural cell populations, as well as increasing the proliferation of subgranular cells in the hippocampal dentate gyrus. 44 The removal of CXCR5 was reported to alleviate sepsis-induced deficits in hippocampal neurogenesis and increase cognitive function in mice in a SAE model. In this context, CXCR5 may act through inflammatory signaling to explain hippocampal neural stem cell proliferation and differentiation, reducing the protection of mature neurons. 32 Sepsis was also reported to deplete the radial pool of glia, like stem cells in the hippocampus. 45 Sepsis may lead to the depletion of the neural stem cell pool and increased gliogenesis in the dentate gyrus. Accordingly, chemokines such as CXCR5 can modulate neurogenesis in hippocampal stem cells in the pathology of sepsis.
A murine model of LPS-induced neuroinflammation showed that the CXCR2 antagonist reduced the transfer rates of neutrophils to the brain. The results suggested that there may be potential chemokines to compensate for chemotaxis for neutrophils to pass through the endothelial barrier associated with reduced neutrophil infiltration. 46 Additionally, IL-8 can cause cytoskeletal reorganization between adjacent endothelial cells by the CXCR2-dependent stimulation of Rac signaling. 47 So, CXCR2 was proposed as a potential pharmacological target for blood-brain barrier (BBB) integrity concerning neuroinflammation in sepsis. 46 A negative correlation was determined between CXCR2 and protein phosphatase 2C delta (Wip1) during sepsis in human neutrophils under in vitro conditions. The pharmacological suppression of Wip1 by the related antagonist can prevent the uptake of CXCR2 in an LPS model of human neutrophils. Accordingly, Wip1 can negatively regulate neutrophil migration and anti-microbial immunity during sepsis. 48 Further investigation of the suppression of Wip1 and chemokine subfamilies such as CXCR2 may be a potential therapeutic target for the treatment of sepsis. Besides, the efficacy of chemokines and cytokines CXCR2 to BBB in sepsis with modulators such as the Rac signaling pathway should be determined in detail with new studies.
CXCR2 expression decreased in CLP-induced sepsis in C57BL/6 mice, and the protection of neutrophil CXCR2 was reduced by the suppression of inducible nitric oxide (iNOS). The downregulation of CXCR2 may also mediate the downregulation of the nitric oxide (NO) receptor associated with the disruption of neutrophil migration observed in severe sepsis. 49 Therefore, the ability of NO to react with superoxide-forming peroxynitrite is important in understanding sepsis.
The peroxynitrite scavenger is involved in the prevention of vascular dysfunction in sepsis. 50 Thus, the efficacy of peroxynitrite in the CXCR2 chemokine-mediated NO signaling pathway may affect vascular function in sepsis. Angiotensin-(1–7) treatment was demonstrated to inhibit LPS-induced organ damage, IL-6 and NO production in rats. Additionally, angiotensin-(1–7) was reported to reduce elevated tissue levels of CXCL1 in endotoxemic rats. The nuclear factor kappa B (NF-kB) signaling pathway appears to mediate the protective effects of Ang-(1–7) in this model. 51 In this sense, NO may lead to vascularization in sepsis by modulating different chemokine families such as CXCR2 and CXCL1. Molecules such as Ang-(1–7) that inhibit NO production are considered a target as a potential therapeutic agent in the treatment of sepsis.
The platelet factor 4 (PF4), a megakaryocyte-derived chemokine involved in innate immunity, may stimulate macrophages. A study in 45 sepsis patients also evaluated the immune response mediated by PF4. The number of platelet-derived microparticles (PMP) carrying PF4 was reported to increase in septic patients. Likewise, increased anti-PF4/heparin antibodies were found in bacterial sepsis patients. 52 The level of circulating PMPs can be increased in various pathological conditions associated with inflammation and sepsis.53,54 In the presence of bacterial infections, PF4 was observed to bind to polyanionic sequences on the surface of aerobic bacteria to form an antigenic complex that induces early antibody formation. 55 In relation to this, a high PF4 + PMP count in sepsis patients may be associated with secondary platelet stimulation due to bacterial infection. 56 The PF4 chemokine in sepsis as a mediator of an innate immune response may be involved in a signaling mechanism mediated by immunoglobulins.
Hence, chemokines, one of the inflammatory mediators, appear to be related to clinical and experimental sepsis pathology via signaling pathways and molecules. Increased chemokine levels in sepsis patients may be potential candidates for diagnostic evaluation. Moreover, the potential of this change observed in patients to affect BBB and hippocampal neural stem cell neurogenesis may be involved in the mechanism of multi-organ damage caused by sepsis. Molecules such as Ang-(1–7) and Wip1 may provide an important therapeutic approach for patients in the treatment of sepsis by modulating the structures affected by chemokines.
Discussion
Summaries of findings reporting the role of inflammatory mediators in sepsis.

TNF-α, tumor necrosis factor-alpha; IL-10, interleukin 10; i.p, intraperitoneal; IL-5, interleukin 5; LPS, lipopolysaccharide; CLP, cecal ligation, and puncture; LTP, Long-term potentiation; WT, Wild type; IL-16, interleukin 16; Ang-(1–7), Angiotensin (1–7); NO, nitric oxide; IFN-γ, interferon-gamma; GSH, Glutathione; MDA, Malondialdehyde; iNOS, inducible nitric oxide; TLR4, toll-like receptor 4; HK-2, proximal tubular epithelial; mir-20a, micro-RNA 20a; PF4, Platelet factor 4; IL-1β, interleukin 1 beta; IL-2, interleukin 2; STAT3, Signal transducer and activator of transcription 3; ERK1/2, Extracellular signal-regulated kinases 1/2; Akt1/2/3, serine/threonine-protein kinase; PMP, PF4-bearing platelet microparticles; CCL2, C-C Motif Chemokine Ligand 2; CCL1, C-C motif chemokine ligand 1; CCL8, C-C motif chemokine ligand 8; CCL20, C-C motif chemokine ligand 20; Trx-1, Thioredoxin-1.
In the acute phase of sepsis, microglia cells can be stimulated by inflammatory mediators and neurotransmitters, causing neuronal dysfunction in the brain. 57 In the pathology of sepsis, cytokines such as IL–10 can affect microglia support cells in the brain. IL-10 prevents the overstimulation of pathological microglia by peripheral endotoxin. In a study of LPS-induced endotoxin in C57BL/6J mice, IL-10 receptor-deficient mice were seen to exhibit neuronal impairment. On the other hand, the addition of microglial TNF-α deficiency improved neuronal disorders. Similarly, IL-10 detection is critical in the prevention of microglia overstimulation during septic shock or bacteremia. Additionally, investigating the gene-level factors that suppress microglial IL-10 production in cases of sepsis or bacteremia may contribute to the understanding of the mechanism. 58
MicroRNAs (miRNAs) acting on the gene level have a diagnostic potential in the pathology of sepsis. 59 The downregulation of microRNA 1184 (miR-1184) could regulate the LPS-induced inflammatory response by suppressing IL-16 in monocytes in a neonatal sepsis model. In 72 cases with neonatal sepsis, IL-16 was shown to be the target of miR-1184, and a negative correlation was found between these parameters. Hence, reduced serum miR-1184 levels may be a potential diagnostic biomarker in neonatal sepsis cases. 60 In another study, miR-150 was defined as a plasma prognostic marker in sepsis patients. Again, in the same study, the plasma levels of TNF-α, IL-10, and IL-18 with sequence complementarity to miR-150 were negatively correlated with the plasma levels of miRNA. miRNA-20a was reported to target CXCL12 and reduce LPS-induced proximal tubular epithelial (HK-2) cell damage via NF-κB and ERK1/2 signaling. In a study that utilized an in vitro model of acute kidney injury with LPS in HK-2 cells, CXCL12 was identified as a direct target of miR-20a by luciferase reporter gene analysis 61. In this respect, the modulation of inflammatory mediators such as TNF-α, IL-10, and IL-18 of the miRNA family, also known as gene-level regulators, may be an important regulatory stage in sepsis. The potential regulatory effect of miRNAs associated with inflammatory mediators might help in reducing the clinical course of sepsis.
Cytokine expressions were evaluated in the epithelium in response to endotoxin-induced systemic inflammation in mice. The transcript levels of IL-10, CCL2, CXCL1, CXCL2, and IL-6 in the isolated choroid plexus 4 h after LPS injection were increased in response to systemic inflammation. 62 So, the choroid plexus may form a link between blood-borne pro-inflammatory mediators and the brain during peripheral inflammation. 63 Thus, unlike the brain parenchyma, structures such as the choroid plexus have been proposed to be the main sites of macrophages that first respond to endotoxin-induced systemic inflammation. 62 In sepsis, the production of substances such as CSF affect the inflammatory response occurring in the choroid plexus. The efficacy of CSF production during the development of sepsis in the choroid plexus was investigated.
The increase in CSF permeability in the choroid plexus in a polymicrobial sepsis model rats with CLP was determined to be associated with oxidative damage and reduced stimulation of the antioxidant enzyme superoxide dismutase. In the end, oxidative stress may be an important regulatory mechanism in CSF dysfunction in experimental sepsis. 64 Additionally, the effect of changing cytokine and chemokine levels due to inflammation in the choroid plexus on the CSF structure in sepsis may be worth considering.
Increased IL-16 expression in the cardiomyocytes of rats reported to be modeled for sepsis with LPS or CLP could be reduced by SOD. IL-16 expression may be regulated by oxidative stress levels during sepsis. However, in a mouse model of LPS-induced sepsis, the neutralization of IL-16 was reported to improve cardiac function by reducing the expression of markers of heart damage 33. In a mouse model of sepsis, it was found that increased oxidative stress may be accompanied by increased cardiomyocyte apoptosis. 65 These data have suggested that oxidative stress may be an important cause of cardiomyocyte apoptosis in sepsis. Additionally, the expression of inflammatory and lipid mediators such as IL-16 may modulate sepsis-induced cardiac injury. Maresin 1, which is a pro-solvent lipid mediator, was reported to have potent regulatory effects on oxidative stress and inflammation in a study about heart damage induced by sepsis in an LPS-induced sepsis model. Maresin 1 improved cardiac function and increased serum SOD, GSH, and Nrf-2 levels in mice in an LPS-induced sepsis model. 66 Regulatory lipid molecules in inflammatory response may also affect heart damage in sepsis through the oxidative stress mechanism.
Differential responses to immune-stimulant drugs were investigated by immunophenotyping peripheral blood mononuclear cells in septic shock patients. An increased expression of leukocyte antigen-DR (HLA-DR) in hyper-response immunity was observed under in vitro conditions in peripheral blood mononuclear cells (PBMC) in septic shock patients. Additionally, IL-17a production by CD8 + T cells was seen to result in an increase in the hyper subgroup. Immunostimulatory therapy could be ineffective or potentially harmful in the hyper-immune response group. Ex vivo stimulations in monitoring responses to immunomodulatory therapies such as interferon-gamma (IFN-γ), immune checkpoint inhibitors, tocilizumab, and steroids may allow the clinician to tailor treatments for sepsis.
67
These stimulations may have a potential in the diagnosis and prognosis of sepsis based on the use of cytokines, chemokines and growth factors. A study in 80 sepsis patients also reported significant increases in IL-6, IL-8, IL-15, and CCL11 levels. Besides, IL-1α and IL-4 levels were low in the cluster 2 patient group associated with unfavorable prognosis or immune dysregulation.
68
In this context, lower IL-4 level
In summary, it may be possible to modulate cytokine and chemokine inflammatory mediators through various signal pathways and stimuli such as NF-κB and ERK1/2. Different degrees of immune responses observed in sepsis patients may be related to these mediators. Moreover, inflammatory mediators regulate resistance to anti-inflammatory drugs administered to reduce sepsis-related complications. Detailed studies are needed to use interleukins and chemokines in prognostic clinical applications in sepsis.
Conclusion
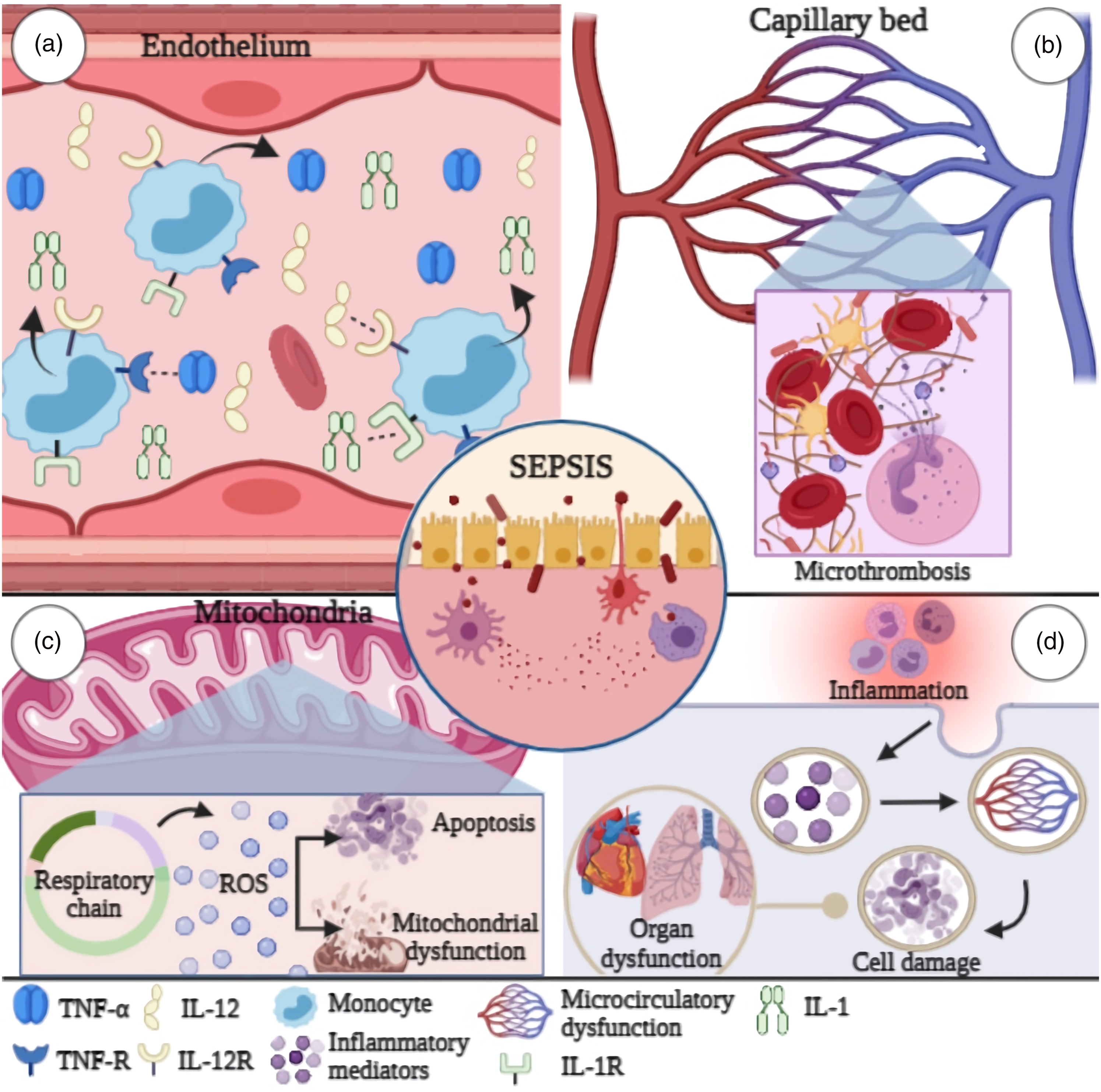
Instead of the devastating clinical picture in sepsis, inflammatory mediators can regulate the inflammatory response. Inflammatory mediators may be regulated by complex signaling networks in the pathogenesis of sepsis. Additionally, secreted by various cells such as neutrophils, inflammatory mediators increase inflammation in target organs. In this respect, pro- and anti-inflammatory mediators in sepsis are also associated with various lethal conditions such as multi-organ failure, vessel damage and pathologies in the brain. New therapeutic approaches to these mediators may help reduce the high mortality rate of sepsis.
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Figures were prepared by biorender application
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.