
Abstract
Introduction
Alzheimer’s disease (AD) affects tens of millions of elderly individuals worldwide and impairs their cognitive and daily functions. 1 AD was first discovered by Dr Alois Alzheimer, and accumulated amyloid plaques were detected in autopsied brains of patients with AD. These amyloid plaques are mainly composed of amyloid-β (Aβ) deposits, which can lead to neuronal dysfunction and even apoptosis. 2 Moreover, Aβ deposits can induce inflammatory reactions in neural cells, and it has been confirmed that cytokines such as interleukin-1β can worsen the prognosis of AD. 3 Accordingly, there are distinct therapeutic targets associated with the Aβ protein, such as targeting amyloid transport and amyloid-based vaccines. 1 Additionally, attenuating Aβ-induced neuronal inflammation and apoptosis is a promising strategy against AD. For instance, rutin reportedly decreases Aβ oligomer levels and neuroinflammation, 4 and cannabidiol can reduce Aβ-triggered neuroinflammation. 5 Moreover, the principal forms of Aβ peptides in AD are Aβ (1–40) and Aβ (1–42). Recently, several studies have highlighted that Aβ (25–35) is highly toxic to neurons.6–8 This peptide can induce neuronal oxidative stress, promote p53 phosphorylation to activate apoptosis, and insert into lipid bilayer cell membrane to damage cell structure.9–12 Yet, Aβ (25–35) has oligomeric, fibrillar, and unaggregated forms, and each form has different biologic function. In this study, we focused on oligomeric Aβ (25–35) and it was employed to model AD in vitro and in vivo.
Panax notoginseng, also called Sanqi, has been adopted as an herbal medicine in China. 13 It contains a diverse range of bioactive saponins, which reportedly prevent atherosclerosis and exert anti-cancer effects. 14 Notably, notoginsenoside R1, a key saponin in P. notoginseng, can suppress metastasis of colorectal cancer, 15 reduce organ ischemia-reperfusion injury, 16 and protect against neuronal damage from amyloid β.17,18 It is worth mentioning that ginsenoside Rg1 can inhibit neural apoptosis and improve hippocampal long-term potentiation and memory in AD.19,20 Although there is much research into notoginsenoside R1 and ginsenoside Rg1, their chemical congener-notoginsenoside R2 has received little attention. Therefore, we aimed to investigate the effects of notoginsenoside R2 on AD. It has also been shown to regulate biological behaviors epigenetically, for instance, miR-30c-5p, which can suppress oxidative stress in cardiomyocytes. 21
Reportedly, miR-27a can mediate various biological effects; for example, it acts as an oncogene in gastric cancer. 22 In neuroscientific research, miR-27a was found to regulate myelination and remyelination as well as protect against neuronal apoptosis by downregulating FOXA3-mediated autophagy. 23 Nevertheless, research exploring the biological function of miR-27a in AD remains scarce. Thus, we aimed to explore if miR-27a has any influence in the pathogenesis of AD. SOX8 is a high mobility group-box transcription factor that is crucial for the early development of embryos, particularly in gender determination. 24 Recent reports have revealed that SOX8 may be involved in mouse brain development and can reprogram fibroblasts into neurons.25,26
In this study, we observed that notoginsenoside R2 improved memory function in AD and inhibited Aβ25-35-induced neuronal apoptosis. Furthermore, notoginsenoside R2 downregulated miR-27a expression, and miR-27a negatively regulated SOX8. Therefore, notoginsenoside R2 could upregulate SOX8 expression and could be used to treat AD in the future.
Methods
Cell culture and proliferation assay
Primary rat cortical neurons were isolated from Sprague–Dawley rats as described earlier.27–29 In brief, neonatal brain tissues were collected, and the cortex was minced and then digested by trypsin for 20 min at 37°C. After that, cells were seeded on 6-well plates coated with poly-L-lysine at the density of 1 × 106 cells/ml. At this stage, they were cultured in Dulbecco’s modified eagle’s medium/F12 (DMEM/F12, Invitrogen, USA) containing 10% fetal bovine serum (Gibco, USA) for 24 h at 37°C with 5% CO2. Then, they were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Beyotime, Shanghai, China) containing 2% B27 (Invitrogen, USA) and 10 μM of cytosine-β-D-arabinofuranoside for about 7 days. Finally, cell viability was assessed by trypan blue staining, and these cells were cultured in RPMI-1640 medium containing 2% B12 and 10% fetal bovine serum at 37°C with 5% CO2. Additionally, notoginsenoside R2 was purchased from MedChemExpress, USA, and dissolved in dimethyl sulfoxide (DMSO).
Quantitative real-time PCR
Primer sequence.

Note: GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
Preparation of Aβ25-35
Amyloid beta-peptide 25–35 (Aβ25-35) was procured from MedChemExpress. According to the manufacturer’s instruction, Aβ25-35 was initially dissolved in cold hexafluoro-2-propanol (HFIP) and then incubated at 24°C for 2 h. Following evaporation to remove HFIP, the resulting film was dissolved in DMSO and diluted in DMEM/F12. Then, the solution was age 48 h at 4°C. After centrifugation at 14,000 g for 10 min at 4°C, most soluble oligomers were in the supernatant and the supernatant was collected for further experiments. We used dot blotting and Western blotting to identify the characteristics of Aβ25-35.
Flow cytometry
After digestion, the cells were resuspended in binding buffer (1 × 105 cells) with Annexin V-Fluorescein isothiocyanate and propidium iodide (Dead Cell Apoptosis Kit, Invitrogen, USA) for 10 min, in accordance with the manufacturer’s instructions. Next, samples were loaded onto a FACSCaliburTM Flow Cytometer (BD Biosciences).
Western blotting
Protein samples were obtained from lysates of primary rat cortical neurons and mouse brain tissue samples using RIPA lysis buffer (Beyotechnology, Shanghai, China). To measure protein expression levels, these samples were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and were then transferred onto polyvinylidene difluoride membranes (Millipore, USA). After blocking with 5% non-fat milk at 4°C for 1 h, the membranes were incubated with anti-cleaved caspase-3 (1:1000, ab231289, Abcam, UK), anti-COX-2 (1:1000, ab179800, Abcam, UK), anti-SOX8 (1:1000, ab226983, Abcam, UK), anti-β-catenin (1:1000, ab68183, Abcam, UK), anti-lamin B1 (1:1000, ab16048, Abcam, UK), and GAPDH (1:1000, ab8245, Abcam, UK) at 4°C overnight, followed by incubation with the appropriate secondary antibody at room temperature for 1 h. Glyceraldehyde 3-phosphate dehydrogenase was used as the endogenous control. Protein blots were visualized using an ECL chemiluminescence kit (Beyotechnology, Shanghai, China).
Dual-luciferase reporter assays
Briefly, primary rat cortical neurons were seeded in 96-well plates. On reaching 60% confluence, the cells were transfected or co-transfected with SOX8-3’UTR-WT, SOX8-3’UTR-MUT, miR-27a mimic, miR-27a mimic negative control, miR-27a inhibitor, and miR-27a inhibitor negative control. After 48 h, luciferase activity was measured in accordance with the manufacturer’s instructions (Promega, USA).
Animal experiments
This research was approved by The First Affiliated Hospital of Guangxi University of Chinese Medicine and the Ethical Code was 2019-003-02. Animals were bought from our animal center.
In total, 40 male senescence-accelerated mice (SAMP8; 20–30 g; 3-month-old),10 normal aging control mice (SAMR1), 40 Sprague–Dawley rats aged between 18 and 20 months old (male, 200–220 g) and 10 Sprague–Dawley rats aged between 3 and 6 months old were purchased from Shanghai SLAC Laboratory Animal Co, Ltd The experimental animals were maintained in our animal laboratory (temperature of 24 ± 1°C, relative humidity of 60 ± 10%, and 12-h light–dark cycle), with free access to food and water.
After one week of adaptation, mice were randomly assigned into four groups (n = 10): the SAMR1 control group (CNG), the SAMP8 AD group, the AD + notoginsenoside R2 (250 mg/kg) co-treatment group, and the AD + miR-27a antagomir (500 mg/kg) co-treatment group. Notoginsenoside R2 and miR-27a antagomir were given to these mice intraperitoneally at 9:00 a.m. every day, and these treatments last 20 consecutive weeks.
In addition, to further validate the role of notoginsenoside, Aβ25-35-induced rat AD model was established as previously reported. 30 Ten Sprague–Dawley rats aged between 3 and 6 months old were allocated into healthy control group. The other 40 Sprague–Dawley rats aged between 18 and 20 months old were randomly categorized into four groups (n = 10): the Sham control group, Aβ25-35-treated group, Aβ25-35 + notoginsenoside R2 (250 mg/kg) co-treatment group, and Aβ25-35 + miR-27a antagomir (500 mg/kg) co-treatment group. To model Aβ25-35-induced AD, rats were initially anesthetized with 1% phenobarbital and then 10 μg of Aβ25-35 (2 μg/μL) were injected into each bilateral hippocampus based on their stereotaxic coordinates at a constant rate within 2 min. The Sham control group was given the same amount of normal saline. After modeling for 8 weeks, Aβ25-35-induced rat AD model was created, and they were given notoginsenoside R2 or miR-27a antagomir intraperitoneally at 9:00 a.m. daily for 20 consecutive weeks. When animal experiments were finished, these rats were sacrificed via the intraperitoneal administration of pentobarbital (90 mg/kg), and rodents were dissected to collect brain tissue.
Behavioral assessment
We performed a step-down passive avoidance test to assess the learning and memory abilities of mice, 3 days after rat AD model was constructed. A continuous electrical stimulation (36 V) was connected to the floor of platform reaction box (10 × 10 × 5 cm) which was separated into two parts by a copper gate. We placed a rubber pad on the right rear corner of each box as a safe area to prevent animals from getting electrocuted. Before assessment, a training process was conducted. During the training process, rats were placed on the platform reaction box for 3 min with 36 V alternating voltage at the bottom of the copper gate, and we recorded the time rats took to react and jump to the pad (reaction time) and counted the number of electric shocks they received within 5 min (error frequency). After 24 h, rats were again placed on the platform again for 3 min, and then placed on the rubber pad. To finish memory training, we recorded the first time when they jumped off the rubber pad (latent period) and counted the number of electric shocks they received within 5 min (error frequency).
Hematoxylin-eosin staining and terminal deoxynucleotidyl transferase dUTP nick end labeling staining
In brief, 4% paraformaldehyde was used for fixing mouse brain tissue for 24 h. The hippocampus was embedded in paraffin, and 5 µm-thick sections were obtained. Following H&E staining, tissue sections were observed under a microscope. Apoptosis was measured using a transferase dUTP nick end labeling (TUNEL) Assay Kit (Invitrogen, USA). Images were analyzed by ImagePro Plus 6.
Statistical analysis
SPSS 20.0 software (IBM Corp., Armonk, NY, USA) was used for data analysis. Data values are presented as the means ± SD from three independent experiments. Differences among multiple groups were examined by analysis of variance (ANOVA), followed by Dunnett’s post hoc test or two-way ANOVA with Bonferroni’s post-tests. Two-tailed p < 0.05 was established as the threshold for statistical significance.
Results
Notoginsenoside R2 reduces apoptosis and inflammation in Aβ25-35-treated neurons
To simulate AD in vitro, different concentrations of Aβ25-35 were used for treating primary rat cortical neurons. Based on flow cytometric observations, it was confirmed that Aβ25-35 induced apoptosis (Figure 1(a)). Thus, 40 μM Aβ25-35 was selected to perform the subsequent experiments. Different concentrations of notoginsenoside R2 were added to Aβ25-35-treated cells, and we observed that notoginsenoside R2 decreased cell apoptosis in a dose-dependent manner. Therefore, 30 μM notoginsenoside R2 was selected for further treatment of primary rat cortical neurons. Additionally, Aβ25-35 treatment increased the protein expression of cleaved caspase-3, and notoginsenoside R2 reversed this effect (Figure 1(b)). Moreover, following Aβ25-35 treatment, the protein expression level of cyclooxygenase 2 (COX-2) increased significantly; notoginsenoside R2 decreased the COX-2 protein expression levels in Aβ25-35-treated primary rat cortical neurons. Thus, the apoptosis and inflammation induced by Aβ25-35 could be attenuated by notoginsenoside R2.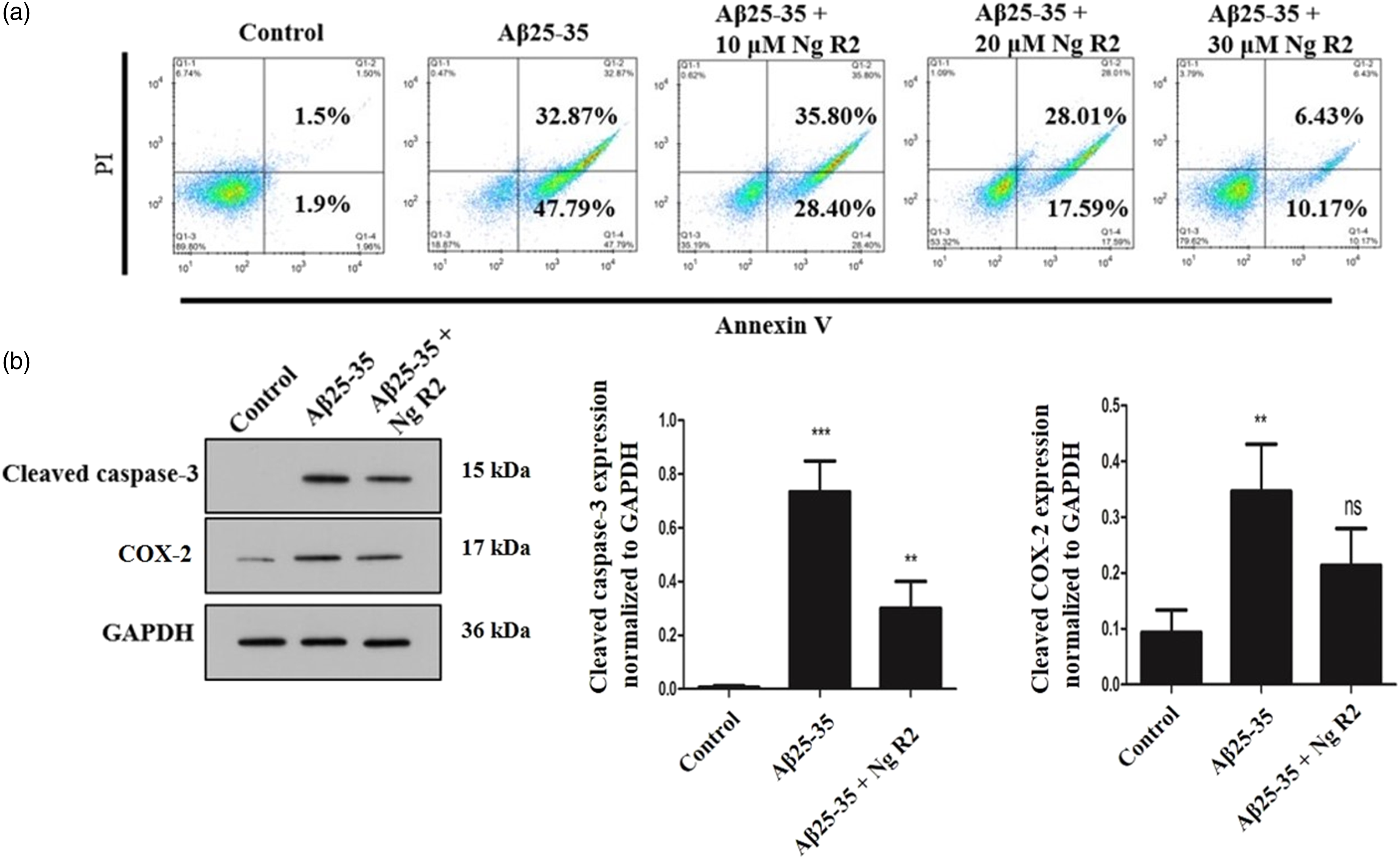
Notoginsenoside R2 downregulates miR-27a to mediate its anti-apoptotic and anti-inflammatory effects in primary rat cortical neurons
Quantitative real-time PCR (qRT-PCR) revealed that notoginsenoside R2 considerably downregulated miR-27a expression (Figure 2(a)). Thus, primary rat cortical neurons were transfected with an miR-27a inhibitor to downregulate miR-27a expression (Figure 2(b)). Next, primary rat cortical neurons were subjected to Aβ25-35 treatment. Results indicated that the downregulation of miR-27a expression alleviated the protein expression levels of cleaved caspase-3 and COX-2 (Figure 2(c)). This finding was corroborated by the flow cytometry results (Figure 2(d)). Therefore, miR-27a might regulate AD.
Notoginsenoside R2 activates SOX8 to mediate its anti-apoptotic and anti-inflammatory effects in primary rat cortical neurons
To elucidate the mechanism underlying the regulation of neuronal apoptosis and inflammation mediated by notoginsenoside R2, qRT-PCR was performed to screen potential molecules. Aβ25-35 treatment downregulated SOX8 mRNA expression, and notoginsenoside R2 upregulated SOX8 mRNA expression (Figure 3(a)). Additionally, at the level of protein expression, SOX8 and β-catenin activities were suppressed following Aβ25-35 treatment; notoginsenoside R2 activated SOX8 and β-catenin activities (Figure 3(b) and (c)). To confirm the role of SOX8 in AD, a SOX8-overexpressing vector was used to transfect primary rat cortical neurons, and SOX8 mRNA expression levels were markedly increased (Figure 3(d)). Accordingly, SOX8 might exert protective effects against AD.
MiR-27a targets SOX8 directly
To explore the relationship between miR-27a and SOX8, DIANA Tools was employed to predict the binding sequences between these two genes (Figure 4(a)). Next, a dual-luciferase reporter assay was performed to confirm this interaction. Primary rat cortical neurons were transfected with pGL3-SOX8-WT luciferase construct and miR-27a mimic, mimic negative (NC), miR-27a inhibitor, or inhibitor negative control (NC). The co-transfection of the pGL3-SOX8-WT luciferase construct and miR-27a mimic downregulated luciferase activity, whereas co-transfection of the pGL3-SOX8-WT luciferase construct and miR-27a inhibitor upregulated luciferase activity (Figure 4(b)). Simultaneously, cells were transfected with the pGL3-SOX8-MUT luciferase construct and miR-27a mimic, mimic negative (NC), miR-27a inhibitor, or inhibitor NC. Co-transfection of the pGL3-SOX8-MUT luciferase construct and miR-27a mimic or miR-27a inhibitor did not influence luciferase activity (Figure 4(c)). Thus, miR-27a could be directly upregulated by SOX8, indicating that miR-27a negatively regulates SOX8.
Notoginsenoside R2 improves cognitive function of AD mice
Notoginsenoside R2 improves the cognitive function of SAMP8 rats.

Note: SAMP8: senescence-accelerated mice P8; AD: Alzheimer’s disease.
N = 10; *p < 0.05, **p < 0.01, ***p < 0.001 by two-sided Student’s t test, versus the control group.
Notoginsenoside R2 improves the cognitive function of Aβ25-35-induced rats.

N = 10; *p < 0.05, **p < 0.01, ***p < 0.001 by two-sided Student’s t test, versus the control group.
Notoginsenoside R2 inhibits apoptosis in AD.
We first established AD model by using SAMP8 rats. The pathological changes in the hippocampus of SAMP8 mice were evaluated by H&E staining. Compared with the control group, the AD group showed sparsely and disorderly arranged neurons, with substantially decreased pyramidal cells, and nuclei stained deep blue (Figure 5(a)). Moreover, notoginsenoside R2 or miR-27a antagomir treatment alleviated the disorganization and abnormal structure of the hippocampus (Figure 5(a)). Apoptosis was evaluated by TUNEL staining. Compared with the control group, an increased number of TUNEL-positive cells was observed in the AD group. Both the AD + notoginsenoside R2 co-treatment group and the AD + miR-27a antagomir co-treatment group displayed fewer TUNEL-positive cells when compared with the AD group (Figure 5(b)). In the meantime, Aβ25-35 was injected into rats to induce AD in vivo. As is illustrated in Figure 5(c), HE staining shows that notoginsenoside R2 or miR-27a antagomir treatment could restore the structure of hippocampus. Also, notoginsenoside R2 or miR-27a antagomir treatment could reduce the apoptosis of neurons induced by Aβ25-35 (Figure 5(d)). Therefore, notoginsenoside R2 attenuated apoptosis in AD via miR-27a in vivo.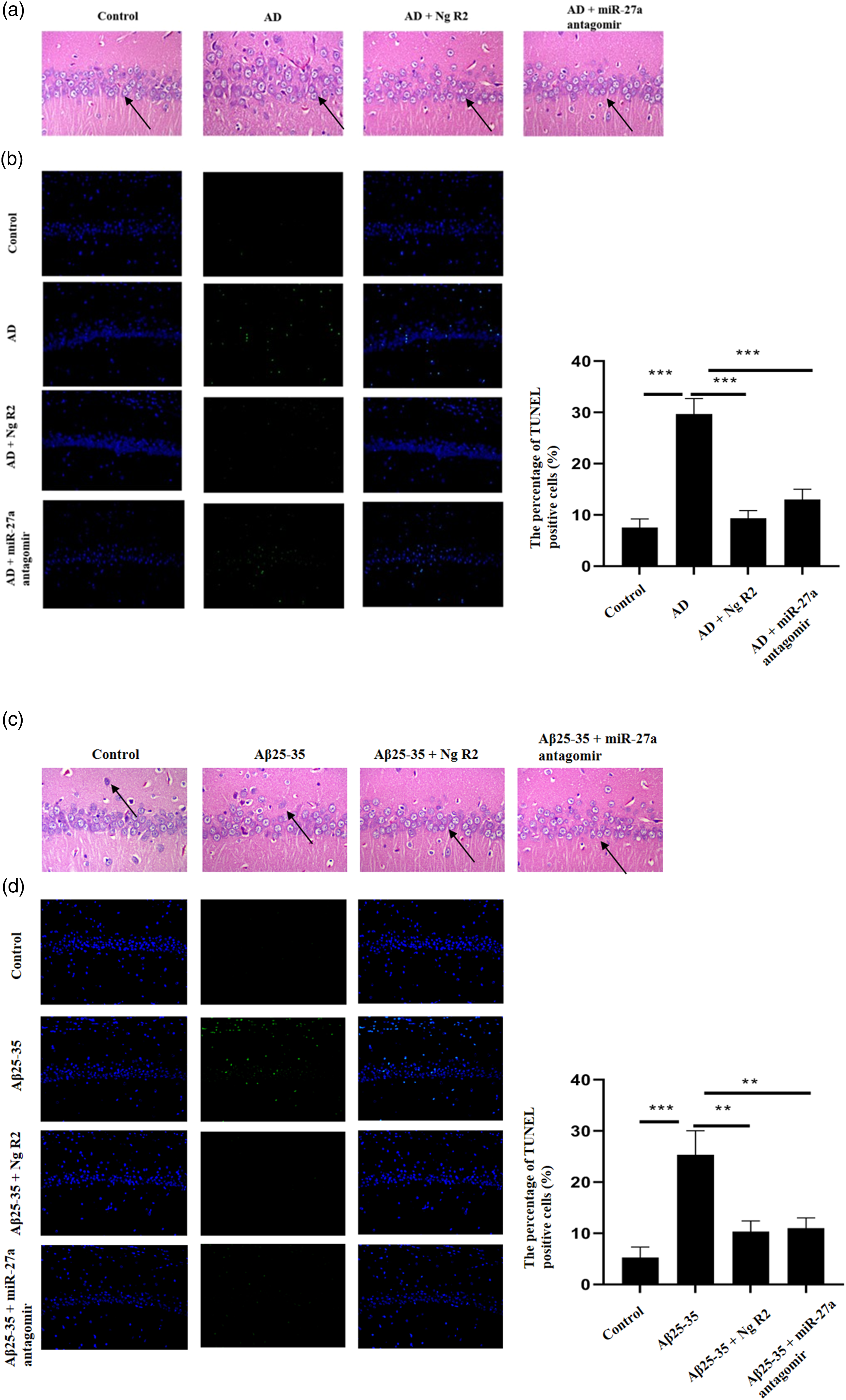
Notoginsenoside R2 reduces apoptosis and inflammation via the miR-27a/SOX8 axis in AD.
To validate our findings in vitro, western blotting was performed. Compared with the control group, the AD group revealed higher expression levels of cleaved caspase-3 and COX-2, with lower expression levels of SOX8 and β-catenin (Figure 6(a)). Additionally, compared with the AD group, the AD + notoginsenoside R2 co-treatment group and the AD + miR-27a antagomir co-treatment group displayed lower expression levels of cleaved caspase-3 and COX-2, with higher SOX8 and β-catenin expression levels. Likewise, Aβ25-35 was injected into rats to induce AD in vivo. Following Aβ25-35 treatment, the expression of cleaved caspase-3 and COX-2 was increased, whereas notoginsenoside R2 or miR-27a antagomir treatment reduced their expression (Figure 6(b)). Moreover, Aβ25-35 treatment downregulated the activity of SOX8 and β-catenin, and notoginsenoside R2 or miR-27a antagomir treatment restored their activity (Figure 6(b)). Hence, notoginsenoside R2 decreased apoptosis and inflammation via the miR-27a/SOX8 axis.
Discussion
AD is the most common form of dementia, with several identified risk factors such as hypertension, high cholesterol, diabetes mellitus, and smoking. 31 In AD, pathological analysis has revealed the occurrence of reduced cerebral blood flow, Aβ deposition, alteration in the blood–brain barrier, degeneration of cholinergic neurons, as well as reactive oxygen species.31,32 Of these changes, accumulation of Aβ peptides is thought to be the underlying cause of AD. In the brain, aberrant Aβ protein deposition can result in diffuse plaques and even infiltrate cerebral vessels, thus increasing the risk of cerebral hemorrhage. 33 Aβ25-35 is one component of the Aβ protein and can cause neurotoxicity and inflammation by activating mitogen-activated protein kinases (MAPK)/nuclear factor κB (NF-κB) and phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling.34,35 Therefore, in this study, Aβ 25–25 was used to modulate neuronal activity.
Notoginsenoside R2 significantly reduced Aβ25-35-induced neuronal apoptosis and inflammation, which was mediated by miR-27a. First, notoginsenoside R2 decreased the expression level of miR-27a. Second, upregulation of this miRNA reversed Aβ25-35-induced neuronal apoptosis and inflammation. Current research mainly focuses on P. notoginseng, and investigations regarding saponins that can improve the prognosis of AD are lacking. It has been revealed that P. notoginseng saponins could enhance the cognitive function of AD rats, reportedly inhibiting the degradation of acetylcholine, reducing neuronal calcium overload, suppressing the activity of NF-κB and cyclooxygenase 2, and decreasing the formation of neurofibrillary tangles. 36 In this study, we demonstrated that notoginsenoside R2 is a potent agent against AD, which is distinct from findings in previous research.
In addition, it was observed that notoginsenoside R2 can activate SOX8 and β-catenin via miR-27a. SOX8 is a sex-determining gene and its mutation results in anomalies of the reproductive system. In tongue squamous carcinoma, SOX8 has been shown to activate β-catenin. 37 In this study, we additionally observed that SOX8 activated β-catenin. Activating Wnt/β-catenin signaling has been recognized as a promising strategy to treat AD, as this signaling can not only regulate synaptic plasticity but also decrease the generation of amyloid-β and tau protein hyperphosphorylation. 38 Moreover, it can reduce oxidative stress in the brain. 39 Thus, notoginsenoside R2 is a promising candidate for the treatment of AD.
However, there are limitations to this research. First, our study utilized a rat cell line rather than a neuronal cell line. Second, although we demonstrated the protective effects of notoginsenoside R2 in AD mouse models, this agent may fail to demonstrate similar efficacy in patients with AD. Third, we need to procure clinical AD samples and analyze relevant genetic expression to confirm our findings. Fourth, the toxicity of Aβ is only one theory to explain the pathogenesis of AD, and that our in vivo data supports that notoginsenoside R2 has protective effects on AD rat model. Our in vivo experiments could not prove that notoginsenoside R2 mediates its effects via inhibiting Aβ-induced apoptosis or inflammation. Thus, this requires further research.
Conclusions
In conclusion, we demonstrated that notoginsenoside R2 can inhibit Aβ25-35-triggered neuronal apoptosis and neuronal inflammation through miR-27a/SOX8/β-catenin signaling. These findings provide novel insights that could be valuable in the field of AD therapeutics.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Project of National Natural Science Foundation of China (No. 81760847, 82060844), the Key Project of Guangxi Natural Science Foundation (No. 2018GXNSFDA050018), Guangxi Medical and Health Appropriate Technology Development and Application Project (No. S2019020), Open Topics of Guangxi University of Chinese Medicine (No. 2019XK018, 2019XK021), the Project of Guangxi Key Laboratory of Chinese Medicine Foundation Research (No. 19-245-14-05), the High-level Talent Team Cultivation Project of Qihuang Project of Guangxi University of Chinese Medicine (No. 2018003), Academic Team of The First Affiliated Hospital of Guangxi University of Chinese Medicine (No. 2018 (146)).