
Abstract
Objective
TGF-β1-induced excessive deposition of extracellular matrix (ECM) and epithelial-mesenchymal transition (EMT) process of tubular epithelial cells play critical roles in the progression of renal fibrosis. We are aimed to explore the effects of lysine-specific demethylase 1 (LSD1) in TGF-β1-treated HK-2 cells and in rats with unilateral ureteral obstruction (UUO), and to investigate the underlying molecular mechanism.
Methods
TGF-β1-treated HK-2 cells and UUO-treated rats were used to establish the model of renal fibrosis in vitro and in vivo, respectively. Protein expression of LSD1, E-cadherin, a-smooth muscle actin (a-SMA), Vimentin, Jagged-1, Notch-1 and Notch-2 were detected by Western blot. The concentrations of type I collagen (Col-I) and Fibronectin (FN) were measured by ELISA. Transwell assay were used to assess cell invasion.
Results
LSD1 was dramatically increased in TGF-β1-stimulated HK-2 cells. Knockdown of LSD1 decreased the TGF-β1-induced secretion of Col-I and FN, and suppressed TGF-β1-induced expression of E-cadherin,α-SMA and Vimentin, while suppressed cell invasion. Consistent with the in vitro data, the severe histopathological damage, collagen deposition and reduced E-cadherin, increased α-SMA induced by UUO was abated by the knockdown of LSD1 in vivo. Moreover, knockdown of LSD1 suppressed TGF-β1-induced expression of Jagged-1, Notch-1 and Notch-2. Furthermore, we found that inhibition of Notch signaling by a γ-secretase inhibitor RO4929097 almost recapitulated the effects of LSD1 knockdown in TGF-β1-induced HK-2 cells, and at least in part reversed the effects of LSD1 overexpression on EMT and ECM deposition in HK-2 cells.
Conclusions
Taken together, LSD1 significantly impact on the progression of TGF-β1-mediated EMT and ECM deposition in HK-2 cells, and it may represent novel target for the prevention strategies of renal fibrosis.
Keywords
Introduction
Renal fibrosis is featured by an excessive deposition of extracellular matrix (ECM) that occurs in chronic kidney disease. Accumulated epithelial–mesenchymal transition (EMT) of renal tubular epithelial cells has been found to contribute to the generation of the matrix-producing cells. 1 Transforming growth factor-β (TGF-β) is one important factor that plays a central role in EMT, which can activate canonical (Smad) and non-canonical (non-Smad) pathways to regulate fibrotic process. 2 Progressive renal fibrosis may lead to the development of end-stage renal failure. 3 Therefore, targeting TGF-β1-induced EMT and ECM deposition may provide more implications for renal fibrosis therapy.
The lysine-specific demethylase 1 (LSD1) is identified as the first histone demethylase. LSD1 has emerged as an epigenetic developmental molecule through the demethylation of histone 3 lysine 4 and histone 3 lysine 9. 4 Through interacting with various partners, such as transcription factors, protein complexes, non-histone substrates, and non-coding RNAs, 5 LSD1 regulates a variety of biological processes, including embryonic development, 6 cellular differentiation, 7 hematopoiesis, 8 spermatogenesis, 9 adipogenesis, 10 and EMT. 11 It has been reported that LSD1 is usually overexpressed in solid cancers and leukemia.12-14 Aberrant expression of LSD1 has been reported to correlate with a more aggressive behavior and poor clinical outcome in different cancers.15,16 In the present study, we speculated that LSD1 might be involved in the progression of renal fibrosis through modulating ECM deposition, EMT, and cell invasion.
Beside the Smad-based pathway,17,18 several non-Smad pathways stimulated by TGF-β1 have been shown to play an important role in the development of renal fibrosis.19,20 It had been reported that the Notch signaling pathway is deeply involved in renal development. 21 The elements of the Notch signaling pathway, such as Jagged-1, Notch-1, and Notch-2, have been identified as TGFβ1-responsive genes.22,23 It has been suggested that the Jagged/Notch pathway may mediate fibrogenic properties of TGF-β.24,25 Furthermore, TGF-β-induced EMT could be blocked by Jagged-1 silencing or by Notch inactivation.23,24 Therefore, we speculated that whether the effects of LSD1 in TGF-β1-induced renal fibrosis were related with the regulation of Jagged-1/Notch signaling.
In the present study, we aimed to determine the expression and effects of LSD1 in TGF-β1-induced HK-2 cells and to investigate the underlying molecular mechanisms.
Material and methods
Cell culture and treatment
HK-2 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultivated in the DMEM/F12 medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) in a 5% CO2 incubator at 37°C. For TGF-β1 treatment, the cells were treated with 2.5, 5, 7.5, and 10 ng/mL TGF-β1 (PeproTech, Rocky Hill, NJ, USA) as indicated time. For RO4929097 treatment, the cells were treated with 1 μM RO4929097 (Selleckchem, Houston, TX, USA) for 24 h before TGF-β1 treatment or cell transfection.
Cell transfection
LSD1 siRNAs (si-LSD1) and corresponding negative control siRNAs (si-NC) were synthesized by RiboBio (Guangzhou, China). PcDNA3.1-LSD1 plasmids were constructed by RiboBio. Cells transfection was performed by Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Western blot assay
Total cellular proteins were extracted. Proteins were separated by SDS-PAGE and transferred to PVDF membranes (Amersham, Buckinghamshire, UK). After blocking overnight with 5% non-fat dried milk, the membranes were then incubated with primary antibody: anti-LSD1, anti-E-cadherin, anti-α-SMA, anti-Vimentin, anti-Jagged-1, anti-Notch-1, anti-Notch-2, or anti-GAPDH (Sigma, San Francisco, CA, USA) overnight at 4°C. Then, the membranes were incubated with corresponding second antibodies (ZSGBBIO, Beijing, China) for 2 h.
Enzyme-linked immunosorbent assay (ELISA)
After different treatments for 48 h, the supernatants of the cells were collected. The concentrations of type I collagen (Col-I) and fibronectin (FN) were measured by the human Col-I and FN ELISA kits (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions.
Transwell assay
After different treatments for 48 h, the cells were harvested. Cells (2 x 105) were suspended in 200 µl serum-free medium and seeded into the Transwell inserts coated with Matrigel (BD Biosciences, USA). Then, 500 µl medium containing 20% FBS was added to the bottoms of the wells. After 24 h incubation at 37°C and 5% CO2, the cells in the upper chamber were removed with cotton swabs. Then, the filter membrane was fixed with 4% paraformaldehyde, and stained with 0.4% crystal violet stain solution. Last, the invasive cells were counted under a light microscope from five randomly selected fields.
Immunofluorescence
Cells grown on cover glass were fixed and treated with 0.1% Triton X-100. After blocking with 0.1% BSA, the cells were incubated with anti-E-cadherin or anti-α-SMA overnight at 4°C. Then, cells were washed and incubated with Alexa Fluor 488 (excitation wavelength: 488 nm; emission wavelength: 518 nm) or Alexa Fluor 594 (excitation wavelength: 594 nm; emission wavelength: 614 nm)-tagged second antibodies (Yeasen Biotechnology, shanghai, China). Additionally, 0.5 µg/ml DAPI (excitation wavelength: 360 nm; emission wavelength: 461 nm) was used to stain nuclei. The slides were then visualized using a fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Animal experiments
Sprague-Dawley (SD) male rats (weighing 200 ± 220 g) were purchased from the Experimental Animal Center of Chengdu DOSSY Experimental Animals CO., LTD (Chengdu, China) and housed in specific pathogen-free environment with a controlled temperature (24 ± 1°C) and 12 h light–dark cycle and had free access to water and standard chow. All animal experimental procedures were carried out according to protocols approved by the Institutional Animal Ethics Committee of Sichuan Province Forestry Center Hospital.
To evaluate the effect of LSD1 knockdown on unilateral ureteral obstruction (UUO)-induced renal fibrosis, rats were first anesthetized by intraperitoneal injection of pentobarbital sodium (45 mg/kg) and then intraperitoneally administered with lentivector negative control (LV-si-NC) or LV-sh-LSD1 as previously described. 26 Five days later, after anesthesia, the UUO surgery was carried out by creating an incision in the left flank and ligating both the proximal and distal left ureter with a 4-0 silk suture. 27 Rats in the sham operation group underwent the same surgical procedure, but the ureter was not ligated.
On day 7 after surgery, the mice were euthanized by intraperitoneal injection of pentobarbital sodium (800 mg/kg), and kidney tissues were harvested for subsequent analyses. The kidney tissues were fixed with 10% formalin, embedded in paraffin, and sectioned into 4 μm. Then, hematoxylin–eosin (H&E) staining, Masson staining, and immunohistochemistry were performed to evaluate the histopathological damage, collagen deposition, and ECM protein expression, respectively.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, USA). All data are presented as the mean ± standard deviation (SD). One-way ANOVA followed by the Bonferroni test was used for multiple group comparisons. p values <0.05 were considered statistically significant.
Results
Expression of LSD1 in TGF-β1-induced HK-2 cells and the interference efficiency of three siRNAs
HK-2 cells were treated with different concentrations of TGF-β1 for 24 h, and then the protein expression of LSD1 was detected by western blot assay. As shown in Figure 1(a), compared with the control group, TGF-β1 treatment induced up-regulation of LSD1 in a concentration-dependent manner. Furthermore, we showed that TGF-β1 treatment induced up-regulation of LSD1 in a time-dependent manner (Figure 1(b)). Treatment with 10 ng/mL TGF-β1 for 24 h was used in the following experiments. We then assessed the interference efficiency of three siRNAs targeting LSD1, as shown in Figures 1(c) and (d), compared with the si-NC group, all the three siRNAs could interfere with the mRNA and protein expression of LSD1, and the interference efficiency of si-LSD1-1 was best. Thus, we used si-LSD1-1 in the following experiments. Expression of LSD1 in TGF-β1-induced HK-2 cells and the interference efficiency of three siRNAs. (a) HK-2 cells were treated with 2.5, 5, 7.5, and 10 ng/mL TGF-β1 for 24 h, and then the protein expression of LSD1 was detected by western blot assay. * p < 0.05, compared with the control group. (b) HK-2 cells were treated with 10 ng/mL TGF-β1 for 0, 6, 12, 18, and 24 h, and then the protein expression of LSD1 was detected by western blot assay. * p < 0.05, compared with the 0 h group. HK-2 cells were transfected with the siRNAs against LSD1 (si-LSD) or negative control siRNA (si-NC). The mRNA(c) and protein (d) expressions of LSD1 were detected by qPCR and western blot assay, respectively. * p < 0.05, compared with the si-NC group.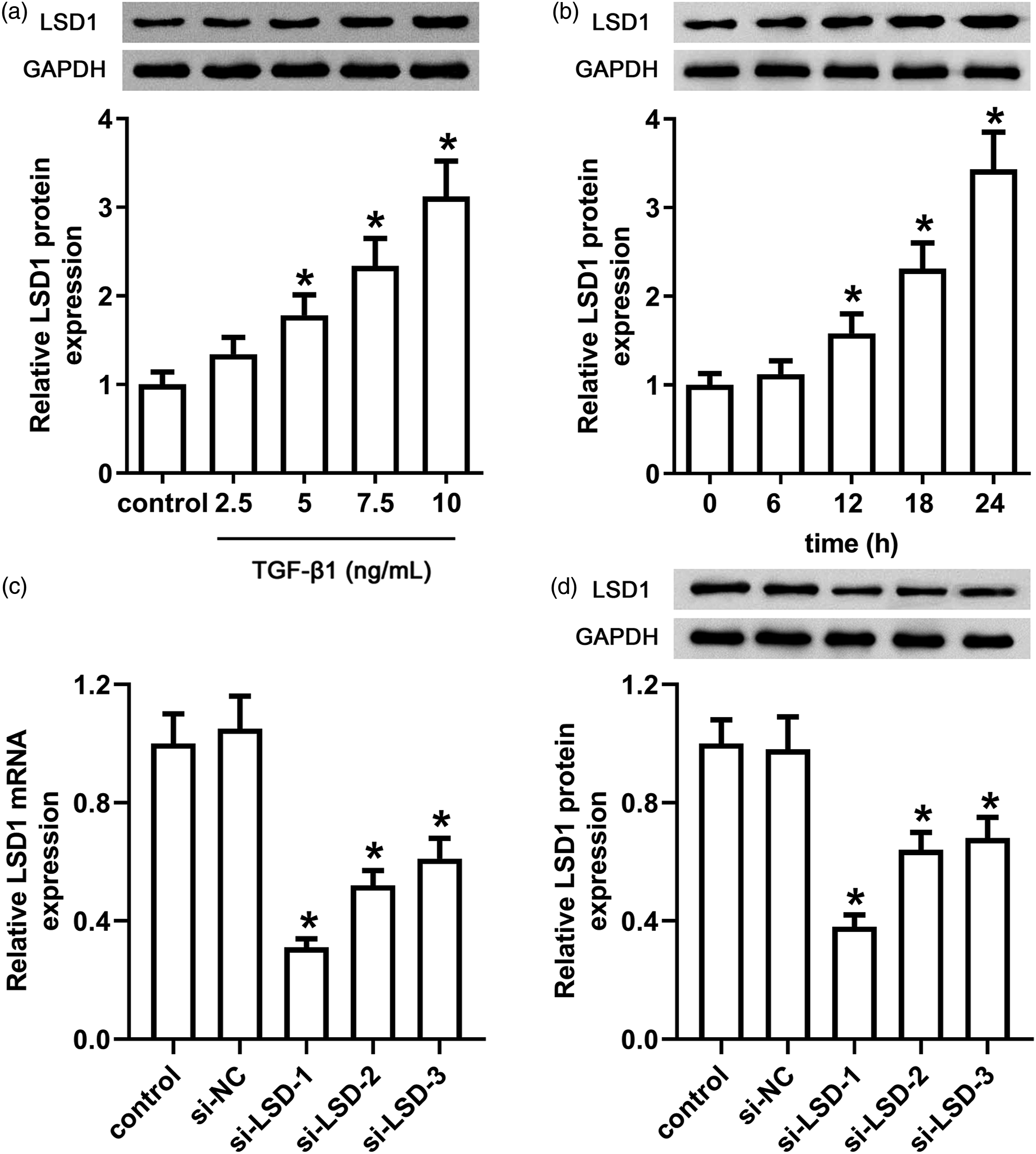
Knockdown of LSD1 suppressed EMT, ECM secretion, and cell invasion in TGF-β1-induced HK-2 cells
To investigate the effects of LSD1 in TGF-β1-induced HK-2 cells, we transfected si-LSD1-1 into HK-2 cells to knockdown the expression of LSD1. As shown in Figure 2(a), compared with the si-NC transfection, the si-LSD1 transfection significantly suppressed its expression in the absence or presence of TGF-β1. We then measured the protein expression of E-cadherin, α-SMA, and Vimentin by western blot assay. As shown in Figures 2(b)‒(e), compared with the si-NC group, the down-regulation of E-cadherin and the up-regulation of α-SMA and Vimentin were found in the si-NC + TGF-β1 group. Compared with the si-NC + TGF-β1 group, the up-regulation of E-cadherin and the down-regulation of α-SMA and Vimentin were found in the si-LSD1 + TGF-β1 group. The similar results about the expression of E-cadherin and α-SMA were found by using immunofluorescence (Figures 2(i) and (j)). Furthermore, we measured the secretion of ECM by ELISA. As shown in Figures 2(f) and (g), compared with the si-NC group, TGF-β1 treatment induced significant elevation of Col-I and FN. Compared with the si-NC + TGF-β1 group, the elevation of Col-I and FN induced by TGF-β1 was blocked by LSD1 knockdown. Then, we assessed the cell invasion by Transwell assay. As shown in Figure 2(h), the invasive cell number in the si-NC + TGF-β1 group was higher than that in the si-NC group. And, the higher cell invasion was significantly suppressed by the knockdown of LSD1. In the absence of TGF-β1, there is no significant difference in the EMT, ECM, and cell invasion between the si-NC group and si-LSD1 group. Knockdown of LSD1 suppressed EMT, ECM secretion, and cell invasion in TGF-β1-induced HK-2 cells. si-NC group, HK-2 cells transfected with si-NC; si-LSD1 group, HK-2 cells transfected with si- LSD1; si-NC + TGF-β1 group, HK-2 cells transfected with si-NC for 24 h, followed by treatment with 10 ng/mL TGF-β1 for 24 h; si-LSD1 + TGF-β1 group, HK-2 cells transfected with si-LSD1 for 24 h, followed by treatment with 10 ng/mL TGF-β1 for 24 h. (a) The protein expression of LSD1 was detected by western blot assay. (b) The protein expression of E-cadherin, α-SMA, and Vimentin was detected by western blot assay. The relative protein expression of E-cadherin (c), α-SMA (d), and Vimentin (e) was shown in histogram. The secretion of Col-I (f) and FN (g) was measured by ELISA. (h) The cell invasion was assessed by Transwell assay, and the invasive cell number was shown in histogram. The expression of E-cadherin (i) and α-SMA (j) was assessed by immunofluorescence assay. * p < 0.05, compared with the si-NC group; # p < 0.05, compared with the si-NC + TGF-β1 group.
Knockdown of LSD1suppressed TGF-β1-induced Jagged-1/Notch signaling
We measured the protein expression of Jagged-1, Notch-1, and Notch-2 by western blot assay. As shown in Figure 3, compared with si-NC group, the up-regulation of Jagged-1, Notch-1, and Notch-2 was found in the si-NC + TGF-β1 group. Compared with the si-NC + TGF-β1 group, the up-regulation of Jagged-1, Notch-1, and Notch-2 induced by TGF-β1 was blocked by LSD1 knockdown. In the absence of TGF-β1, there is no significant difference in the above protein expression between the si-NC group and si-LSD1 group. Knockdown of LSD1 suppressed TGF-β1-induced Jagged-1/Notch signaling. (a) The protein expression of Jagged-1, Notch-1, and Notch-2 was detected by western blot assay. The relative protein expression of Jagged-1 (b), Notch-1 (c), and Notch-2 (d) was shown in histogram. * p < 0.05, compared with the si-NC group; # p < 0.05, compared with the si-NC + TGF-β1 group.
Inhibition of Notch signaling almost recapitulated the effects of LSD1 knockdown
We tried to assess the effects of Notch signaling in TGF-β1-induced HK-2 cells. A γ-secretase/Notch signaling pathway inhibitor RO4929097 was used to suppress Notch signaling. Then, we measured the expression of EMT-associated protein, the secretion of ECM, and the cell invasion. As shown in Figures 4(a)‒(d), compared with the TGF-β1 group, the down-regulation of E-cadherin and up-regulation of α-SMA and Vimentin induced by TGF-β1 were blocked by RO4929097. Furthermore, the elevation of Col-I and FN induced by TGF-β1 was also blocked by RO4929097 (Figures 4(e) and (f)). And, the higher cell invasion induced by TGF-β1 was significantly suppressed by RO4929097 (Figure 4(g)). The above data indicate that Notch signaling was involved in the regulation of TGF-β1-induced HK-2 cells. Considering that knockdown of LSD1 suppressed Notch signaling, we speculated that inhibition of Notch signaling was involved in the function of LSD1 knockdown. Notch signaling was involved in the regulation of TGF-β1-induced HK-2 cells. control group, untreated cells; TGF-β1 group, HK-2 cells treated with 10 ng/mL TGF-β1 for 24 h; RO4929097 + TGF-β1 group, HK-2 cells pre-treated with 1 μM RO4929097 for 24 h, followed by treatment with 10 ng/mL TGF-β1 for 24 h. (a) The protein expression of E-cadherin, α-SMA, and Vimentin was detected by western blot assay. The relative protein expression of E-cadherin (b), α-SMA (c), and Vimentin (d) was shown in histogram. The secretion of Col-I (e) and FN (f) was measured by ELISA. (g) The cell invasion was assessed by Transwell assay, and the invasive cell number was shown in histogram. * p < 0.05, compared with the control group; # p < 0.05, compared with the TGF-β1 group.
Notch signaling mediated the effects of LSD1
To confirm whether Notch signaling was involved in the function of LSD1, LSD1 was overexpressed in HK-2 cells which were treated with RO4929097. As shown in Figures 5(a)‒(d), compared with the control group, LSD1 overexpression induced the down-regulation of E-cadherin and the up-regulation of α-SMA and Vimentin; meanwhile, LSD1 overexpression led to the elevation of Col-I and FN (Figures 5(e) and (f)) and the higher invasive cell number (Figure 5(g)). RO4929097 treatment alone showed no obvious difference compared with the control group, but significantly reversed the effects of LSD1 overexpression, leading to the up-regulation of E-cadherin, the down-regulation of α-SMA and Vimentin, the decrease of Col-I and FN, and the lower invasive cell number, compared with the LSD1 overexpression group. Notch signaling mediated the effects of LSD1. control group, untreated cells; LSD1 group, HK-2 cells transfected with pcDNA3.1-LSD1 plasmids; RO4929097 group, HK-2 cells treated with 1 μM RO4929097 for 24 h; LSD1 + RO4929097 group, HK-2 cells pre-treated with 1 μM RO4929097 for 24 h, followed by transfected with pcDNA3.1-LSD1 plasmids. (a) The protein expression of E-cadherin, α-SMA, and Vimentin was detected by western blot assay. The relative protein expression of E-cadherin (b), α-SMA (c), and Vimentin (d) was shown in histogram. The secretion of Col-I (e) and FN (f) was measured by ELISA. (g) The cell invasion was assessed by Transwell assay, and the invasive cell number was shown in histogram. * p < 0.05, compared with the control group; # p < 0.05, compared with the LSD1 group.
Knockdown of LSD1 attenuates renal fibrosis in rats with UUO
To validate the effects of LSD1 in vivo, we then investigated whether LSD1 knockdown had an anti-renal fibrotic activity in rats with UUO. As shown in Figure 6, as for the sham rats, the HE staining, Masson staining, and immunohistochemistry all showed no obvious differences between the LV-sh-NC and LV-sh-LSD1 groups. Compared with the Sham+LV-sh-NC group, the severe histopathological damage, collagen deposition and reduced E-cadherin, and increased α-SMA were found in the UUO+ LV-sh-NC group, indicating that UUO treatment induced renal fibrosis in the rats. Compared with the UUO+LV-sh-NC group, the histopathological damage and collagen deposition induced by UUO were abated in the UUO+LV-sh-LSD1 group, although the changes were not significant. However, the reduced E-cadherin and increased α-SMA induced by UUO were markedly suppressed in the UUO+LV-sh-LSD1 group. Knockdown of LSD1 attenuates renal fibrosis in rats with UUO. SD rats were intraperitoneally administered with lentivector negative control (LV-si-NC) or LV-sh-LSD1. Five days later, the UUO surgery was carried out by creating an incision in the left flank and ligating both the proximal and distal left ureter with a 4-0 silk suture. Rats in the sham operation group underwent the same surgical procedure, but the ureter was not ligated. Hematoxylin–eosin (H&E) staining (a), Masson staining (b), and immunohistochemistry (c and d) were performed to evaluate the histopathological damage, collagen deposition, E-cadherin and α-SMA protein expression, respectively.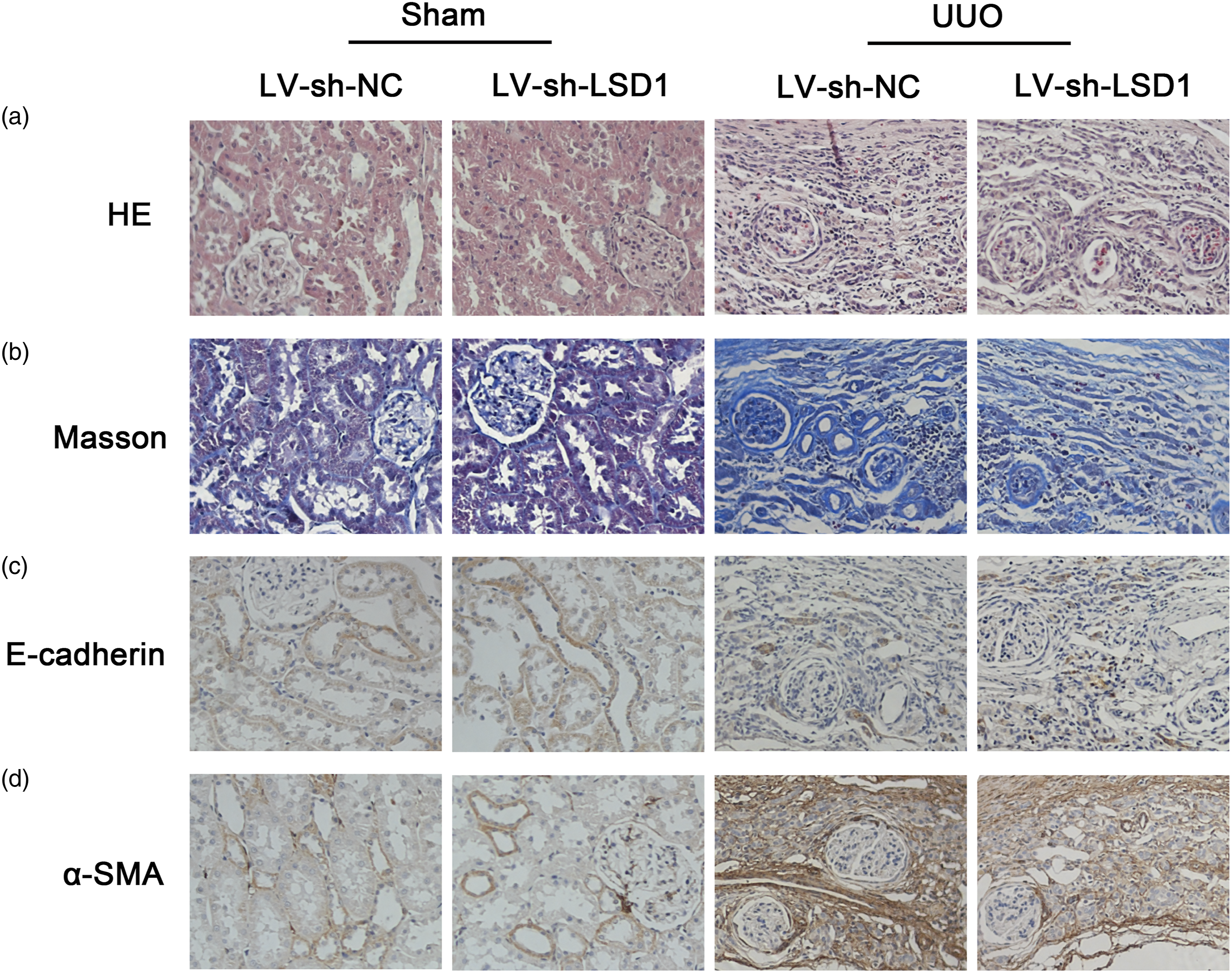
Discussion
During the development of renal fibrosis, TGF-β1 has been identified as the key mediator that drives fibroblast activation and ECM deposition.28-30 Recent therapeutic strategies for renal fibrosis have aimed at antagonizing the fibrogenic action of TGF-β/Smad signaling. In the present study, we showed that LSD1 was dramatically increased in TGF-β1-stimulated HK-2 cells. Knockdown of LSD1 decreased the TGF-β1-induced secretion of Col-I and FN and suppressed TGF-β1-induced expression of E-cadherin, α-SMA, and Vimentin while suppressing cell invasion. Moreover, knockdown of LSD1 suppressed TGF-β1-induced expression of Jagged-1, Notch-1, and Notch-2. Furthermore, we found that inhibition of Notch signaling by a γ-secretase inhibitor RO4929097 almost recapitulated the effects of LSD1 knockdown in TGF-β1-induced HK-2 cells, and at least in part reversed the effects of LSD1 overexpression on EMT and ECM deposition in HK-2 cells.
By using cultured HK-2 cells, we used TGF-β1 to induce an in vitro renal fibroblastic cell model. Then, we showed that TGF-β1 treatment induced up-regulation of LSD1 in HK-2 cells in a concentration and time-dependent manner, suggesting that LSD1 may be deeply involved in the regulation of TGF-β1-induced HK-2 cells. Thus, we transfected si-LSD1 into HK-2 cells to knockdown the expression of LSD1, to further investigate the roles of LSD1. EMT is a process in which epithelial cells lose their polarity and acquire the migratory and invasive characteristics of mesenchymal cells. 31 EMT is driven by a complex transcriptional program, leading to the silencing of epithelial genes (e.g., E-cadherin and Occludin) as well as the activation of mesenchymal genes (e.g., α-SMA, N-cadherin, Vimentin, and FN).11,32,33 The crucial role of LSD1 in EMT and cell invasiveness has been highlighted by multiple studies. And LSD1 depletion reduces EMT-driven cell migration, suggesting that LSD1 is an important regulator in epigenetic reprogramming during EMT.11,34 In the present study, we showed that TGF-β1 treatment induced significant elevation of ECM (Col-I and FN) accumulation. And, the elevation of ECM induced by TGF-β1 was blocked by LSD1 knockdown. Furthermore, TGF-β1 treatment induced significant down-regulation of E-cadherin and up-regulation of α-SMA and Vimentin, which were also blocked by LSD1 knockdown. Meanwhile, the higher invasive cell number in the TGF-β1 group was significantly suppressed by LSD1 knockdown. These data suggest that LSD1 could decrease the ECM deposition, EMT, and cell invasion in TGF-β1-induced HK-2 cells. Consistent with the in vitro data, the severe histopathological damage, collagen deposition and reduced E-cadherin, and increased α-SMA induced by UUO were abated by the knockdown of LSD1 in vivo, indicating that knockdown of LSD1 attenuates renal fibrosis in rats with UUO.
It has been reported that TGF-β1-induced renal fibrosis are frequently mediated by the canonical Smad signaling.35,36 In addition to the Smad-dependent signaling pathway, TGF-β1 also induces different Smad-independent signaling pathways to promote renal fibrosis, 20 including Jagged-1/Notch signaling pathway. Actually, after completion of renal development, Notch signaling is greatly suppressed. 37 When the Notch signaling pathway went on to be over-activated, EMT and fibrosis may be progressed.38,39 Murea et al. 40 have indicated that the reactivation of the Notch signaling pathway in the tubulointerstitium was correlated well with the severity of fibrosis. In the present study, we showed that TGF-β1 treatment induced significant up-regulation of Jagged-1, Notch-1, and Notch-2, and the up-regulation was blocked by LSD1 knockdown, indicating that LSD1 could suppress the activation of TGF-β1-induced Jagged-1/Notch signaling pathway. To further determine whether Notch signaling was involved in the roles of LSD1, we assessed the effects of Notch signaling in TGF-β1-induced HK-2 cells. Pharmacological inhibition of Notch signaling in the TGF-β1-induced HK-2 cells was performed. RO4929097 is a γ-secretase inhibitor, which is required for Notch receptor cleavage, and it has been well tolerated in clinical phase I/II anti-cancer trials.41,42 Suppression of Notch activation by RO4929097 could block the down-regulation of E-cadherin, and up-regulation of α-SMA and Vimentin induced by TGF-β1 or LSD1 overexpression, inhibit the elevation of Col-I and FN and the higher cell invasion induced by TGF-β1 or LSD1 overexpression. The above data indicate that the regulation of Notch signaling was closely associated with the roles of LSD1 on TGF-β1-induced renal fibrosis.
Actually, mitogen-activated protein kinases (MAPKs) pathways, RhoA, and Rac1 small GTPases have all been implicated in TGF-β1-induced Smad-independent signaling pathways mediating TGF-β-induced EMT and renal fibrosis.43-47 Thus, whether the roles of LSD1 on TGF-β1-induced renal fibrosis are depended on other signaling pathways still need more investigations. Moreover, the downstream networks of TGF-β1-mediated signaling in the development of renal fibrosis still need more detailed understanding.
Taken together, our data showed that knockdown of LSD1 decreased the TGF-β1-induced ECM accumulation and suppressed TGF-β1-induced EMT while suppressing cell invasion. Moreover, the effects of LSD1 on TGF-β1-mediated EMT and ECM deposition may be related to the regulation of the Jagged-1/Notch signaling pathway. Our findings elucidate the molecular action of LSD1 in attenuating renal fibrosis, suggesting that LSD1 may be a novel target for the therapeutic strategies to alleviate renal fibrosis.
Footnotes
Acknowledgments
The authors would like to thank members of nephrology department in Sichuan Province Forestry Center Hospital for helpful discussions.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.