
Abstract
Artesunate (ARS) has been shown to be highly effective against chloroquine-resistant malaria. In vitro studies reported that ARS has anticancer effects; however, its detrimental action on cancer cells may also play a role in its toxicity toward normal cells and its potential toxicity has not been sufficiently researched. In this study, we investigated the possible cytotoxic effects using normal BRL-3A and AML12 liver cells. The results showed that ARS dose-dependently inhibited cell proliferation and arrested the G0/G1 phase cell cycle in both BRL-3A and AML12 liver cells. Western blotting demonstrated that ARS induced a significant downregulation of cyclin-dependent kinase-2 (CDK2), CDK4, cyclin D1, and cyclin E1 in various levels and then caused apoptosis when the Bcl-2/Bax ratio decreased. Conversely, the levels of intracellular reactive oxygen species (ROS) were increased. The ROS scavenger N-acetylcysteine can significantly inhibit cell cycle arrest and apoptosis induced by ARS. Thus, the data confirmed that ARS exposure impairs normal liver cell proliferation by inducing G0/G1 cell cycle arrest and apoptosis, and this detrimental action may be associated with intracellular ROS accumulation. Collectively, the possible side effects of ARS on healthy normal cells cannot be neglected when developing therapies.
Introduction
Artesunate (ARS) is a water-soluble semisynthetic derivative of artemisinin. ARS and its active metabolite dihydroartemisinin (DHA) are potent blood schizonticides, highly effective against multidrug-resistant strains of Plasmodium falciparum. The effectiveness of ARS is due to its rapid and complete hydrolysis to form DHA that is three- to fivefold more active than the parent compound. 1 Hence, ARS is the World Health Organization-recommended first-line treatment of severe malaria in endemic countries. Beyond its antimalarial properties, growing evidence reveals that artemisinins have robust inhibitory effects against viruses, inflammation, autoimmune diseases, and cancers, 2 and many studies showed that artemisinins exhibit significant anticancer effects against human tumor cells with minimal effects on normal cells. 3,4 However, the widespread usage of ARS made it imperative to characterize its potential toxicity in in vivo assays. The most frequently reported adverse event is post-ARS delayed hemolysis, and the incidence is approximately 15% of treated malarial patients. 5 In addition, the other major side effects associated with artemisinins are neurological and reproductive toxicity, 6 relative mild hepatic injury, and other manifestations. 5 For example, sub-chronic exposure of ARS at 8 mg/kg/day dose for a 45-day period caused hepatic damage in rats, 7 and a higher dose of 16 mg/kg/day for 7 days led to hepatotoxicity in guinea pigs. 8
Apart from in vivo studies, particular attention should also be paid to the potential toxicity of artemisinins to normal cells in vitro. Several studies reported that the anticancer mechanism of artemisinins mainly works by inducing cell cycle arrest, cell apoptosis, generation of reactive oxygen species (ROS), and other dysfunctions. 3,9 However, the detrimental properties of artemisinins on cancer cells may also play a role in their toxicity toward normal cells. Several studies demonstrated that artemisinin and its analogs have cytotoxic effects on healthy cells, such as normal human bronchial epithelial (HBE) cells 10 and porcine oocytes during maturation in in vitro assays. 11 The liver plays a pivotal role in the metabolism of most drugs and is highly susceptible to drug-induced toxicity. At present, little is known about the toxicity of ARS on normal liver cells. In this study, we investigated the potential toxic effects on normal liver cell lines (BRL-3A and AML12) in vitro by detecting the cell proliferation, cell cycle, and apoptosis and analyzed the relationship between the toxic effects and intracellular ROS generation.
Materials and methods
Chemicals and antibodies
ARS (purity > 98%) was purchased from TCI (Shanghai, China) Development. A Cell Counting Kit-8 (CCK8) was obtained from Dojindo Molecular Technologies (Kumamoto, Japan). Dimethyl sulfoxide (DMSO), N-acetylcysteine (NAC), and 2′,7′-dichlorofluorescin diacetate (DCFH-DA) were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). An FITC Annexin V Apoptosis detection kit was obtained from BD Biosciences (New Jersey, USA). An enhanced chemiluminescence (ECL) kit and polyvinylidene difluoride (PVDF) were acquired from Merck Millipore (Darmstadt, Germany). A bicinchoninic acid (BCA) protein assay kit and cell cycle and apoptosis analysis kit were purchased from Beyotime Institute of Biotechnology (Beijing, China). ARS was dissolved in DMSO and the final concentrations of DMSO did not exceed 0.1%.
The primary antibodies against β-actin (cat no. 4970S), cyclin-dependent kinase-2 (CDK2) (cat no. 2546S), CDK4 (cat no. 12790S), cyclin D1 (cat no. 2922S), cyclin E1 (cat no. 20808S), Bax (cat no. 2772S), and Bcl-2 (cat no. 3498S) were obtained from Cell Signaling Technology (Danvers, Massachusetts, USA). The corresponding secondary antibody (cat no. 111-035-003) was purchased from Jackson ImmunoResearch (West Grove, Pennsylvania, USA).
Cell line and cell culture
The BRL-3A and AML12 cell lines were obtained from the cell bank of Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) or DMEM-F12 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and allowed to adhere in an incubator at 37°C with 5% CO2 and humidified atmosphere of 95% air.
The effect of ARS on cell proliferation by CCK8 assay
The BRL-3A and AML12 cells were seeded into 96-well plates at a density of 6000 cells per well and incubated at 37°C overnight. Then the cells were exposed to ARS at different concentrations for 24 h or 48 h, respectively. Cells treated with blank vehicles (DMSO) were used as a vehicle control. CCK8 was subsequently added to each well and incubated for 2 h. The optical density (OD) was measured at 450 nm using an EMax microplate reader (Molecular Devices, Sunnyvale, California, USA). The OD of the vehicle control group was equivalent to 100% viable cells, and the cell viability was calculated as a percentage of the vehicle control.
Cell cycle analysis
The cell cycle was determined with a Cell Cycle and Apoptosis Analysis kit (Beyotime). The BRL-3A and AML12 cells grown on six-well culture plates were harvested after different treatments. In the analysis of ARS on cell cycle regulatory, the cells were treated with different concentrations of ARS (0, 5, 10, and 20 μM on BRL-3A and 0, 1, 2.5, and 5 μM on AML12 for 24 h). To examine the role of ROS in ARS-induced G0/G1 cell cycle arrest, the BRL-3A cells were co-incubated with or without antioxidant agent NAC and ARS for 24 h. The remaining processing steps were carefully performed according to the manufacturer’s instruction.
Apoptosis analysis
The ability of ARS to induce apoptosis in the BRL-3A cells was determined using an FITC Annexin V Apoptosis detection kit and Western blotting. The BRL-3A cells in six-well plates treated with different concentrations of ARS (0, 5, 10, and 20 μM), respectively, for 36 h were collected. For the flow cytometry analysis, the cells were resuspended using binding buffer and stained with Annexin-V-FITC for 15 min, followed by staining with propidium iodide (PI) for 5 min. All of the stained cells were analyzed immediately by flow cytometry. Apoptosis-related proteins were detected by Western blotting.
Western blotting
The treated cells were lysed in ice-cold RIPA buffer supplemented with protease inhibitor and phosphatase inhibitor (Applygen). After protein quantification with BCA, an equal amount of protein in each sample (20 μg) was separated by electrophoresis using 12% sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred to PVDF membranes. The membranes were blocked with 5% skim milk for 1 h at room temperature and then incubated overnight at 4°C with the primary antibodies. After washing three times with TBST, the membranes were then incubated with anti-goat IgG horseradish peroxidase (HRP) secondary antibody (diluted 1:5000) for 1 h at room temperature. Protein signal bands were determined on a ChemiDoc XRS (Bio-Rad, Marnes-la-Coquette, France) using an ECL kit.
Measurement of ROS
ROS generation by the BRL-3A cells was detected via flow cytometry with a DCFH-DA oxidation-sensitive probe. After the cells were processed for the indicated time, they were resuspended and incubated in PBS containing DCFH-DA (10 μM) for 30 min in the dark at 37°C. After washing twice with phosphate buffered saline (PBS), the cells were resuspended in 0.5 ml PBS and immediately submitted to flow cytometry for analysis. The data were analyzed using FCS Express software (FlowJo, BD Bioscience).
Statistical analyses
All the experimental data were presented as the mean ± standard error of the mean. A one-way analysis of variance with a Duncan’s multiple comparison test was utilized to determine statistical differences among the groups. All statistical analyses were performed using IBM SPSS Statistics 22.0 software, and diagrams were performed by GraphPad Prism (version 5.0) software. Differences were considered statistically significant when p < 0.05 or p < 0.01.
Results
ARS induced proliferation inhibition and G0/G1 phase cell cycle arrest in BRL-3A and AML12 cells
CCK8 assays at 24 and 48 h showed that ARS exposure significantly inhibited the proliferation of both the BRL-3A and AML12 cells in a profound dose- and time-dependent manner, and the AML12 cells were more sensitive to ARS as shown in Figure 1(a). To determine whether cell cycle arrest is involved in anti-proliferation of ARS, the distribution of the cell cycle in the BRL-3A and AML12 cells was determined by flow cytometry and the results suggested that ARS exposure (more than 5 μM) for 24 h caused a marked cell cycle arrest at the G0/G1 phase in the BRL-3A cells (p < 0.01), with a corresponding significant decrease in the number of cells at the S phase (p < 0.01) (Figure 1(b) and (d)). Similar to the results of the BRL-3A, the AML12 cells were also notably arrested at the G0/G1 phase when exposed to more than 2.5 μM of ARS for 24 h (Figure 1(c) and (e)). Then the primary proteins that control the commitment of eukaryotic cells to transition through the G1 phase to enter the DNA synthesis S phase in the BRL-3A cells were examined via Western blotting. The results suggested that more than 5 μM of ARS exposure could markedly decrease the expression of cyclin E1 and cyclin D1 in the BRL-3A cells as shown in Figure 1(f). ARS at higher doses of 10 and 20 μM significantly downregulated the expression of CDK2 and CDK4 in the tested cells. These results revealed that ARS interfered with BRL-3A cell proliferation via downregulating CDK4, cyclin D1, CDK2, and cyclin E1 expression and then induced a cell cycle arrest at the G0/G1 phase.

Effects of ARS on cell viability and cell cycle arrest in BRL-3A and AML12 cells. (a) BRL-3A and AML12 cells were treated with various concentrations of ARS for 24 and 48 h. Data are expressed as the percentage relative to the control group. *p < 0.05 and **p < 0.01 versus control. (b) BRL-3A and (c) AML12 cells were, respectively, treated with different concentrations of ARS for 24 h and subsequently analyzed by flow cytometry. (d) Quantified results from (b). (e) Quantified results from (c). *p < 0.05 and **p < 0.01 represent statistically significant higher versus control, and # p < 0.05 and ## p < 0.01 represent statistically significant lower versus control. (f) BRL-3A cells were treated with ARS (0, 5, 10, and 20 μM) for 24 h and then collected to assess the protein levels of CDK2, cyclin E1, CDK4, and cyclin D1 using Western blotting. *p < 0.05; **p < 0.01. All of the data are shown as mean ± SEM of three independently repeated tests. ARS: artesunate; CDK: cyclin-dependent kinase; SEM: standard error of the mean; NS: not significant.
ARS-induced apoptosis in BRL-3A cells
The experiments certified that ARS inhibits cell proliferation and arrests cell cycle in BRL-3A cell lines. We next examined whether cell apoptosis was involved in the anti-proliferative effects of ARS on the cells. As shown in Figure 2(a) and (b), exposure to different concentrations of ARS for 36 h induced cell apoptosis in a dose-dependent manner, and 10 and 20 μM of ARS exposure markedly induced cell apoptosis (p < 0.05 and p < 0.01). To further estimate cellular apoptosis, the proto-oncoproteins (Bcl-2 and Bax) were detected (Figure 2(c)). Over 10 μM of ARS treatment downregulated the anti-apoptotic protein of Bcl-2 and upregulated the pro-apoptotic proteins of Bax, resulting in a marked reduction in the Bcl-2/Bax ratio in the BRL-3A cells.
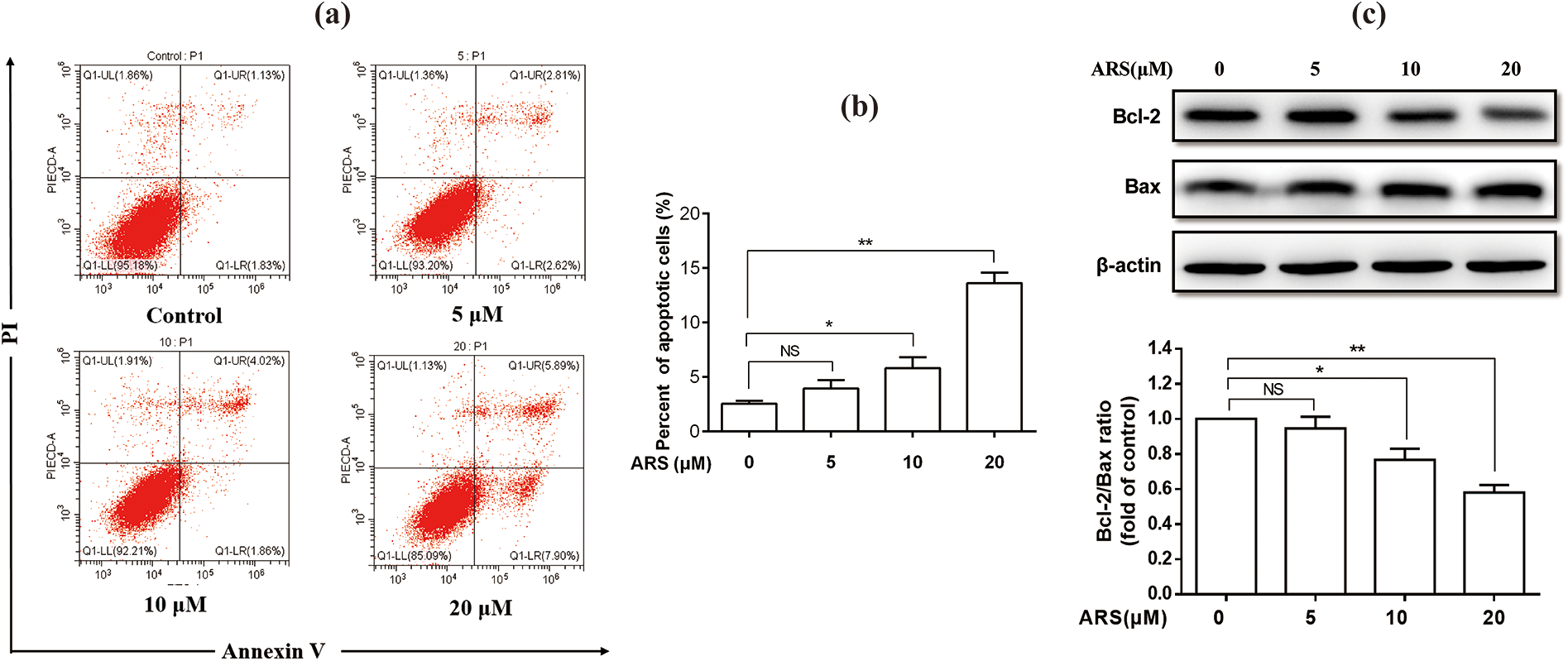
The apoptotic effects of ARS on BRL-3A cells. The cells were treated with 0, 5, 10, and 20 μM of ARS for 36 h to assess the apoptosis. (a) Analyses of apoptosis by flow cytometry and its quantitative results (b). (c) The effect of ARS on the ratio of Bcl-2/Bax was assessed by Western blotting. All of the values are expressed as mean ± SEM (n = 3). *p < 0.05; **p < 0.01. ARS: artesunate; SEM: standard error of the mean; NS: not significant.
ROS is involved in cell cycle arrest in BRL-3A cells
The effect of ARS on ROS generation in BRL-3A cells was investigated. As shown in Figure 3(a) and (b), the generation of ROS gradually increased as the concentration of ARS increased. Only 5 μM of ARS exposure increased the ROS levels in the cells with no statistical significance. However, ARS exposure at doses of 10 and 20 μM significantly enhanced the intracellular ROS accumulation (p < 0.01), and the higher dose, the ROS levels increased. Thus, the data suggest that ARS exposure induced the generation of ROS in the BRL-3A cells in a concentration-dependent manner. To examine whether ROS was involved in ARS-induced G0/G1 cell cycle arrest, the ROS scavenger agent NAC was used in this experiment. The result showed that 20 μM of ARS dramatically arrested the cell cycle in the G0/G1 phase, and this action could be markedly suppressed or prevented by NAC pretreatment (Figure 3(c) and (d)). NAC pretreatment also counteracted the proliferative inhibition effect of ARS on the cells, especially at 40 μM for 24 h and 20 μM for 48 h (p < 0.05) detected by CCK8 assay as shown in Figure 3(e).

ROS is involved in cell cycle arrest in BRL-3A cells. (a) BRL-3A cells were treated with different concentrations of ARS (0, 5, 10, and 20 μM) for 24 h and the level of ROS was determined by flow cytometry. (b) Quantified results from (a). *p < 0.05 and **p < 0.01 versus control group. (c) The cells were treated with 20 μM of ARS and with or without NAC for 24 h. (d) Quantified results from (c). *p < 0.05 and **p < 0.01 versus control, and # p < 0.05 and ## p < 0.01 versus ARS group. (e) BRL-3A cells were treated with different doses of ARS and with or without NAC for 24 and 48 h. *p < 0.05; **p < 0.01. All of the data are shown as mean ± SEM of three independently repeated tests. ROS: reactive oxygen species; ARS: artesunate; NAC: N-acetylcysteine; SEM: standard error of the mean.
Removal of ROS alleviated ARS-induced apoptosis in BRL-3A cells
ARS was shown to induce BRL-3A apoptosis in the previous experiments, so we further investigated whether NAC could inhibit cell apoptosis. The results are shown in Figure 4(a) and (b). The enhanced apoptosis of the BRL-3A by 20 μM of ARS exposure for 36 h was markedly downregulated by NAC pretreatment (Figure 4(c)) (p < 0.05). Moreover, NAC pretreatment relieved the depression of the Bcl-2/Bax ratio. The results suggest that the increase in ROS is involved in ARS-induced apoptosis.
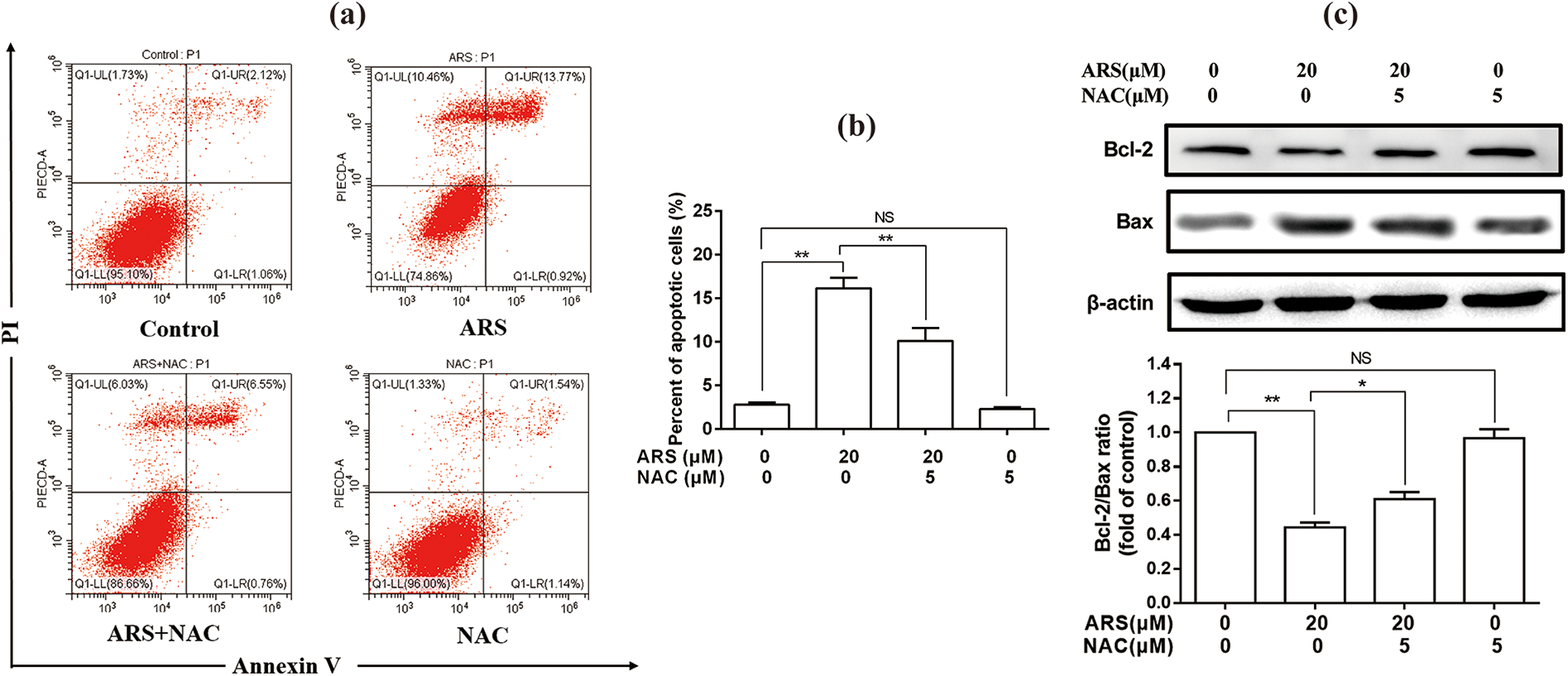
Removal of ROS alleviated ARS-induced apoptosis in BRL-3A cells. The cells were treated with 20 μM of ARS and with or without NAC for 36 h to assess the anti-apoptosis effect of NAC. (a) Apoptosis was analyzed by flow cytometry. (b) The quantitative results of (a). (c) The effect of NAC on ARS-induced downregulated Bcl-2/Bax ratio was assessed by Western blotting. All of the values are expressed as mean ± SEM (n = 3). *p < 0.05; **p < 0.01. ROS: reactive oxygen species; ARS: artesunate; NAC: N-acetylcysteine; SEM: standard error of the mean; NS: not significant.
Discussion
A meta-analysis of clinical trials concluded that artemisinin drugs are considered safe and well tolerated with no difference among the various derivatives for malaria patients even in the second or third trimester of pregnancy. 12 Common side effects were nausea, vomiting, and diarrhea, which are also symptoms of malaria itself. However, animal experiments show considerable toxicity upon application of artemisinin drugs. 13 For example, DHA treatment induced abnormal embryonic phenotypes and promoted embryonic angiogenesis in zebrafish. 14 ARS exposure can cause congenital malformations in rodents and apoptosis of some immune cells. 15 –17 Some in vitro experiments also found that artemisinin, DHA, and artemether treatment led to the death of neuronal cells and glial cells and the transformation of neuronal cells. 18,19 However, most drugs are metabolized and converted to more polar compounds in the liver to facilitate excretion, so the liver is highly susceptible to drug-induced toxicity. Thus, it is important to investigate the potential adverse action of drugs in liver cells.
In this study, we investigated the effect of ARS on rat liver BRL-3A cells and mouse liver AML12 cells. Our results showed that ARS exposure could inhibit cell proliferation in a profound dose-dependent manner. In addition, cycle analysis revealed that ARS dose-dependently induced a marked cell cycle arrest at the G0/G1 phase in the two cell lines, and this action can be directly explained in BRL-3A cells by the downregulation of cell cycle-related proteins of CDK2, CDK4, cyclin D1, and cyclin E1. This action is very similar to its inhibiting roles on tumor cells by affecting the cell cycle at any stage, with the G0/G1 to S phase inhibition most commonly observed, and its mechanism of action is achieved by altering the expression and activity of regulatory enzymes in the cell cycle. 3,9 For example, artemisinin can directly inhibit CDK4 gene expression leading to cell cycle arrest in prostate cancer cells. 20 Similarly, artemisinin induced a G1 cell cycle arrest in cultured human Ishikawa endometrial cancer cells by downregulating CDK2 and CDK4 transcript and protein levels. 21 In the current study, we found that 5 μM of ARS could significantly downregulate the protein expression levels of cyclin D1 and cyclin E1. In addition, 10 μM of ARS dramatically abrogated the expression of CDK2 detected by Western blotting. In another in vitro study, ARS (60 μM) induced cell cycle arrest at the G0/G1 phase through the downregulation of cyclin A1, cyclin B, cyclin D1, CDK2, CDK4, and CDK6 in human epidermoid carcinoma cells. 4 These data indicate that a relatively lower dose of ARS can lead to cell cycle arrest in normal rat liver cells.
Similar to our results, DHA, the main active metabolite of artemisinin, at a dose of 40 μM induced ROS accumulation in porcine oocytes during in vitro maturation and blocked cell cycle progression. 11 Actually, all artemisinin-based drugs were reported to contain the antimalarial chemical group endoperoxide bridge that can generate ROS in malaria parasites. 22 Generally, the anticancer effect of artemisinin drugs is usually achieved by excessive ROS generated by the endoperoxide moiety in cancer cells, then leading to apoptosis and other dysfunctions. 23,24 To better understand the anti-proliferative effect of ARS on BRL-3A cells, we further investigated the apoptotic percentage using flow cytometry. Our study found obvious apoptosis in the BRL-3A cells at 36 h after 10 and 20 μM of ARS exposure (Figure 2). Therefore, we further investigated the apoptosis marker proteins. The results showed that 10 and 20 μM of ARS exposure markedly enhanced the ratio of Bcl-2/Bax in the BRL-3A cells. Moreover, ARS exposure for 24 h dose-dependently caused a higher accumulation of ROS in the tested cells. One recent study reported that HeLa cells, after ARS treatment, generated ROS earlier than when the cytotoxic effects occurred, thus indicating that ROS may initiate factors triggering cell damage. 25
Notably, the enhanced ROS levels after ARS exposure were in line with the inhibition extent of the G1/S transition of the cell cycle triggered by ARS in the current study. In general, ROS are perceived as toxicants that induce various detrimental effects, such as cell dysfunction, death, or malignant transformation. 26 We assume that reducing or inhibiting ROS generation using some antioxidants will prevent or mostly block ARS-induced cell cycle arrest. We further tested the effect of ROS scavenger NAC on the cell cycle and apoptosis in the BRL-3A cells. The result demonstrated that the detrimental role of ARS-induced cell cycle arrest could be significantly inhibited by NAC. Moreover, NAC pretreatment markedly alleviated ARS-induced apoptosis in the BRL-3A cells. Therefore, we speculate that the accumulation of ROS induced by ARS may contribute to cell cycle arrest at the G0/G1 phase and pro-apoptosis in BRL-3A cells.
Regarding the selectivity of the cytotoxic effect of artemisinin drugs on cancer cells with minimal effect on normal tissue and cells, this selectivity is disputable in different studies and the mechanism remains unclear at present. A recent study compared the selectivity of artemisinin-based drugs on human lung normal and cancer cells. The study used five assays to evaluate the cytotoxic effects of artemisinin (250–500 μM) and ARS (100–150 μM) on normal HBE cells and lung adenocarcinoma A549 cells, both cell lines developed from lung tissues, thus compromising the specificity of different types of tissue. The five cytotoxic assays clearly showed that artemisinin drugs caused dose-dependent cytotoxicity in both the HBE and A549 cells with a slight selectivity to the latter. 10 Both demonstrated elevated levels of intracellular ROS and increased DNA damage, and cancer cells were more vulnerable to artemisinin drugs. However, the selectivity of ARS was no more than 3 times, 10 not as high as 36 times more potent in killing malignant Molt-4 cells than its normal counterpart reported in a previous study. 27 Other studies also found that normal cells, such as vascular smooth muscle cells and embryonic erythroblasts, showed the cytotoxic effect of artemisinin (50, 100 μM) and its derivative DHA (7 μM). 28,29 Therefore, the selectivity of artemisinins deserves further research.
In conclusion, our study demonstrated that relatively lower dose ranges of ARS exposure can inhibit the proliferation of BRL-3A and AML12 cells through a marked G0/G1 phase cell cycle arrest in a dose-dependent manner, which was further evidenced by a prominent downregulation of key cell cycle regulatory proteins. ARS exposure can induce apoptosis of BRL-3A. However, ARS-induced cell cycle arrest and apoptosis were significantly inhibited by adding ROS scavenger NAC to the tested cells, indicating ROS accumulation contributes to the anti-proliferative roles and apoptosis of ARS in BRL-3A cells. Combining the previously discussed reports about the cytotoxic effect of artemisinin drugs on normal cells, more attention should be paid to the possible side effects of artemisinin-based drugs on healthy normal cells when developing therapies.
Footnotes
Author contributions
SY, HY, and JL designed the study. SY and HY conducted the flow cytometry and Western blotting experiments. XZ and SW assisted with the CCK8 assay. SY and JL wrote the manuscript. YT, ML, and RB reviewed the manuscript. SY and HY contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the National Natural Science Foundation [Grant Number: 31672595] and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).