
Abstract
Arsenic is an environmental poison and is a grade I human carcinogen that can cause many types of damage to the body. The skin is one of the main target organs of arsenic damage, but the molecular mechanisms underlying arsenic poisoning are not clear. Arsenic is an epigenetic agent. Histone acetylation is one of the earliest covalent modifications to be discovered and is closely related to the occurrence and development of tumors. To investigate the role of acetylated histone H3K18 (H3K18 ac) in arsenic-induced DNA damage, HaCaT cells were exposed to sodium arsenite (NaAsO2) for 24 h. It was found that arsenic induced the downregulation of xeroderma pigmentosum A, D, and F (XPA, XPD, and XPF—nucleotide excision repair (NER)-related genes) expression, as well as histone H3K18 ac expression, and aggravated DNA damage. Chromatin immunoprecipitation quantitative polymerase chain reaction (ChIP-qPCR) analysis showed that H3K18 acetylation in the promoter regions of XPA, XPD, and XPF was downregulated. In addition, the use of the histone deacetylase inhibitor trichostatin A (TSA) partially inhibited arsenic-induced DNA damage, inhibited deacetylation of H3K18 ac in the promoter regions of XPA, XPD, and XPF genes, increased acetylation of H3K18, and promoted the transcriptional expression of NER-related genes. Our study revealed that NaAsO2 induces DNA damage and inhibits the expression of NER-related genes, while TSA increases the H3K18 ac enrichment level and promotes the transcriptional expression of NER, thereby inhibiting DNA damage. These findings provide new ideas for understanding the molecular mechanisms underlying arsenic-induced skin damage.
Keywords
Introduction
Arsenic is a toxic metal substance that can cause many types of damage to human health. The skin is the main target organ of arsenic-induced damage. 1 At present, many studies on the pathogenesis of arsenic-induced damage have been carried out, and many hypotheses regarding this process have been formed, including the induction of oxidative stress, inhibition of DNA repair, interference in signal transduction, and chromosomal abnormalities. However, the exact mechanism underlying arsenic-induced damage has not yet been elucidated. 2,3 Arsenic is an epigenetic poison that can cause changes in a variety of epigenetic regulatory patterns. It has been observed that HaCaT cells exposed to 10 μM sodium arsenite (NaAsO2) exhibit changes in H4R3 and H3R17 methylation levels, suggesting that arsenic can cause alterations in histone modifications. 4
DNA damage and repair disorder is an important mechanism underlying tumorigenesis. Arsenic exposure can interfere with the process of DNA damage and repair in cells, resulting in chromosomal DNA damage. 5 It has been observed that histone modifications are involved in a variety of repair pathways and play an important role in the regulation of DNA repair. 6 Arsenic can inhibit DNA repair enzymes and promote DNA damage. 7 Nucleotide excision repair (NER) is one of the major repair pathways after DNA injury. The proteins xeroderma pigmentosum A, D, and F (XPA, XPD, and XPF) play significant roles in the NER pathway. A study by Wei et al. 8 revealed that arsenic can decrease the messenger RNA (mRNA) and protein expression of XPD in L-02 cells, suggesting that arsenic could lead to DNA damage. Our previous study showed that arsenic exposure altered the levels of histone H3K9me2 modification in peripheral blood lymphocytes, and the level of H3K9me2 modification in samples from the population exposed to arsenic was closely related to the degree of DNA damage. 9 One study found that arsenic can reduce the modification level of H3K36me3 with the occurrence of DNA damage and the destruction of mismatched repair pathways. 10 It has been suggested that histone modifications are involved in the repair process of DNA damage induced by arsenic and its related compounds.
Histone acetylation is closely related to tumorigenesis. 11 It can reduce the affinity between histones and DNA, promoting gene transcription. Previous studies have shown that acetylated histone H3K18 (H3K18 ac) is one of the major histone modifications related to disease. It is involved in the occurrence and development of disease by regulating the expression of genes involved in key pathways, such as the cell cycle and DNA damage repair. 12 –14 A study has shown that histone modifications play an important role in regulating the expression of DNA injury and repair-related factors. 15 Thus, we first explore H3K18 ac has a regulatory effect on NER-related genes during the repair of arsenic-induced DNA damage. Next, the removal of histone acetylation is regulated by histone deacetylase (HDAC). Trichostatin A (TSA) is a HDAC inhibitor that increases the degree of histone acetylation by inhibiting HDAC activity. Overall, H3K18 ac levels are altered in the epithelial cells of patients with asthma, and TSA increases the acetylation level of H3K18. 16
Therefore, we studied the role of H3K18 ac and the NER-related genes XPA, XPD, and XPF in arsenic-induced DNA damage in HaCaT cells. At the same time, TSA was used to alter the level of histone acetylation to explore the role of H3K18 ac in the repair of arsenic-induced DNA damage to investigate the mechanism underlying arsenic-induced DNA damage.
Materials and methods
Cell culture and treatment
Cells from the human keratinocyte line HaCaT were purchased from the Chinese typical Culture Center of Wuhan University. The cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (Gibco, Carlsbad, California, USA) at 37°C in a 5% CO2 incubator. Cells in the logarithmic phase were used in this experiment. NaAsO2 staining solution was prepared using autoclaved double distilled water. The HaCaT cells (1 × 104 cells/well) were inoculated into 96-well plates so that the degree of cell fusion increased from 80% to 90%. HaCaT cells were exposed to 0.00, 2.50, 5.00, 10.00, and 20.00 μmol/L NaAsO2 for 24 h. The proliferation activity of the cells was then observed.
TSA intervention
Inoculating HaCaT cells in a six-well plate increased the fusion degree from 80% to 90%. The cells were treated with 250 nmol/L TSA for 1 h in advance and then with 10 μmol/L NaAsO2 for 24 h. Dimethyl sulfoxide (DMSO) was used for the control group. The cells were divided into the following groups: blank group, 10.00 μmol/L NaAsO2 (hereafter referred to as the arsenic group), solvent control group (DMSO), TSA (250 nmol/L) group, and combined treatment group (250 nmol/L TSA + 10.00 μmol/L NaAsO2).
Single-cell gel electrophoresis test
The cells were washed thrice with phosphate-buffered saline (PBS), fully blown, and collected by digestion with 0.25% trypsin. The level of DNA damage was observed by fluorescence microscopy according to the degree of cell lysis and electrophoretic neutralization and staining procedures. Under the microscope, 100 cells were randomly counted. The number of cells showing the trailing phenomenon was counted, and the positive rate of trailing cells was calculated, which were expressed as the percentage of DNA in the tail (TailDNA%) and the olive tail moment (OTM), respectively.
RNA extraction and quantitative reverse transcription PCR
The cells from each group were washed twice with aseptic precooled PBS. Total RNA was extracted using the conventional Trizol reagent, and 2 μg of each total RNA sample was reverse transcribed into complementary DNA using a reverse transcriptase kit (TaKaRa, Japan). Bio-Rad CFX96 (California, USA) was used for the quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis. The sequences of the primers used are shown in Table S1. After the reaction, the Bio-Rad CFX Manager software 3.1 was used for data analysis according to the detected cycle threshold (Ct value). The Ct values of each sample were corrected relative to the value of β-actin. The expression levels of mRNA were determined based on the 2−ΔΔCt method. 17
Western blots
After the treatments, the cells were washed thrice with a precooled phosphate buffer. Next, the total protein was extracted. The target protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel electrophoresis and transferred to a polyvinylidene fluoride membrane with 300 mA constant flow for 60 min on ice. The membranes were then blocked using 5% skimmed milk powder after being sealed at room temperature for 2 h. Anti-β-actin (Abcam, United Kingdom) (1:1000), anti-H3K18 ac (Abcam) (1:1000), anti-XPA (Abcam) (1:1000), anti-XPD (Abcam) (1:1500), and anti-XPF antibodies (Abcam) (1:2500) were diluted using the primary antibody diluent, and the membranes were incubated with the primary antibodies on a shaking table at 4°C overnight. Goat antirabbit horseradish peroxidase (HRP) conjugate (1:10,000, Proteintech, Wuhan, China) was used as a secondary antibody. For quantitative analysis, western blots were analyzed with a ChemiDoc XRS + Imaging System (Bio-Rad, USA). The signal for each protein was normalized to actin detected on the same blot, and ratios with average control values were determined.
ChIP assay
This experiment was performed using the EZ-Magna chromatin immunoprecipitation (ChIP™) A/G ChIP kit (Millipore, Upstate, USA) and the EZ-Zyme Chromatin Prep kit (Millipore, USA). A total of 4 × 106 cells were collected from each treatment group, and EZ-Zyme enzyme cocktail containing 1% protease inhibitor was used to lyse the cells. The intracellular DNA was digested using the EZ-Zyme enzymatic cocktail II, and the protein-DNA complexes were immunoprecipitated using anti-H3K18 ac antibodies. Immunoglobulin G (IgG) was used as the control. Protein-binding magnetic beads were used to enrich the DNA-protein complexes. These mixtures were washed sequentially with low-salt wash solution, high-salt wash solution, lithium chloride salt wash solution, and Tris-EDTA (TE) buffer. The protein mixtures were then incubated with protease K solution at 62°C for 2 h. ChIP-eluting buffer was used to elute the immunoprecipitation complexes, and purified DNA was obtained for quantitative polymerase chain reaction (qPCR) analysis. H3K18 ac is mainly enriched in the promoter region of the genes and is related to their transcriptional activation. 18 We designed two pairs of primers (ChIP1 and ChIP2) for the promoter regions of the XPA, XPD, and XPF genes; the sequences of the primers used are shown in Table S2. The results were expressed as a ratio of the immunoprecipitated DNA to the input DNA.
Statistical analyses
All the experimental data were calculated and expressed as the means ± standard deviations and analyzed using SPSS 21.0 statistical software. The data were analyzed using the normality test and homogeneity of variance test. One-way analysis of variance was used for the comparison of data between groups, and linear regression analysis was used for the correlation analysis. The level of significance was set to α = 0.05.
Results
Arsenic induces DNA damage and decreases the expression of the XPA, XPD, and XPF genes in HaCaT cells
The main target organ of arsenic and its derivative compounds is the skin. Human immortalized skin keratinocytes (HaCaT cells) are derived from normal adult skin keratinocytes, which are similar to normal skin cells and can be passaged for a long time. The HaCaT cell line is frequently used to study the mechanism of arsenic toxicity. 19 The results of the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay showed that the proliferation of HaCaT cells increased slightly following treatment with 1.25 μmol/L NaAsO2 and then decreased with increasing arsenic doses. The 50% inhibitory concentration of NaAsO2 for HaCaT cells was 18.95 μmol/L (Figure 1(a)). In this study, the cells were exposed to arsenic doses of 0.00, 2.50, 5.00, and 10.00 μmol/L, and the exposure time was 24 h. DNA damage in cells was detected by the single-cell gel electrophoresis method. The results showed that the TailDNA% and OTM values increased significantly with increasing arsenic doses. The data showed that arsenic caused DNA damage and decreased the cell survival rate (Figure 1(b) and (c)). The qRT-PCR analysis revealed that the mRNA transcription levels of the XPA, XPD, and XPF genes decreased to varying degrees with the increase in arsenic (Figure 1(d)). Western blotting analysis showed that the expression of XPD and XPF proteins also decreased gradually, however, arsenic cannot change the protein expression of XPA (Figure 1(e) and (f)). These data suggest that arsenic can decrease the expression of the XPA, XPD, and XPF, and can aggravate DNA damage in cells, which may be one of the important causes of arsenic-induced DNA damage.

NaAsO2 induces DNA damage and decreases the transcription levels of XPA, XPD, and XPF mRNA and the expression of the XPD and XPF proteins in HaCaT cells. The survival rate of HaCaT cells was measured after 24 h. (a) The MTT assay was used to examine the survival rate of HaCaT cells (mean ± SD, n = 6). (b) DNA damage was detected by SCGE (Ethidium bromide staining, 200×). (c) The degree of DNA damage was calculated based on the OTM and TailDNA% values (mean ± SD, n = 100). (d) The relative expression levels of XPA, XPD, and XPF were detected by qRT-PCR (mean ± SD, n = 3). (e) Western blotting and (f) relative protein expression levels of XPA, XPD, and XPF (mean ± SD, n = 3). *p < 0.05, compared with the control group. SCGE: single-cell gel electrophoresis; OTM: olive tail moment; SD: standard deviation; qRT-PCR: quantitative reverse transcription-polymerase chain reaction; NaAsO2: sodium arsenite; XPA: xeroderma pigmentosum A; XPD: xeroderma pigmentosum D; XPF: xeroderma pigmentosum F.
Arsenic can reduce the modification level of H3K18 ac and its enrichment level in the promoter regions of the XPA, XPD, and XPF genes
With the development of epigenetics studies, an increasing number of in vitro experimental results have suggested that arsenic induces changes at the level of specific histone modifications in cells. 20 –22 Next, we investigated whether H3K18 ac could regulate the expression of NER-related genes. We found that after HaCaT cells were exposed to different doses of NaAsO2, the levels of H3K18 ac modification decreased with increasing arsenic concentration (Figure 2(a) and (b)). ChIP is a technique used to detect the accumulation of histones in DNA using a specific histone-labeled biological antibody while maintaining the combination of histones and DNA. 23 Histone acetylation can influence DNA damage repair by mediating chromatin relaxation. Chromatin immunoprecipitation quantitative polymerase chain reaction (ChIP-qPCR) was used to detect the H3K18 ac enrichment level in the promoter regions of the XPA, XPD, and XPF genes. Compared to the cells from the control group, the H3K18 ac enrichment levels in the ChIP1 region and ChIP2 region of the XPA, XPD, and XPF gene promoters decreased significantly in the cells treated with arsenic (Figure 2(c)). These results suggest that arsenic can reduce the levels of H3K18 ac enrichment in the promoter regions of NER-related genes, inhibit gene transcription, and promote the occurrence of DNA damage.
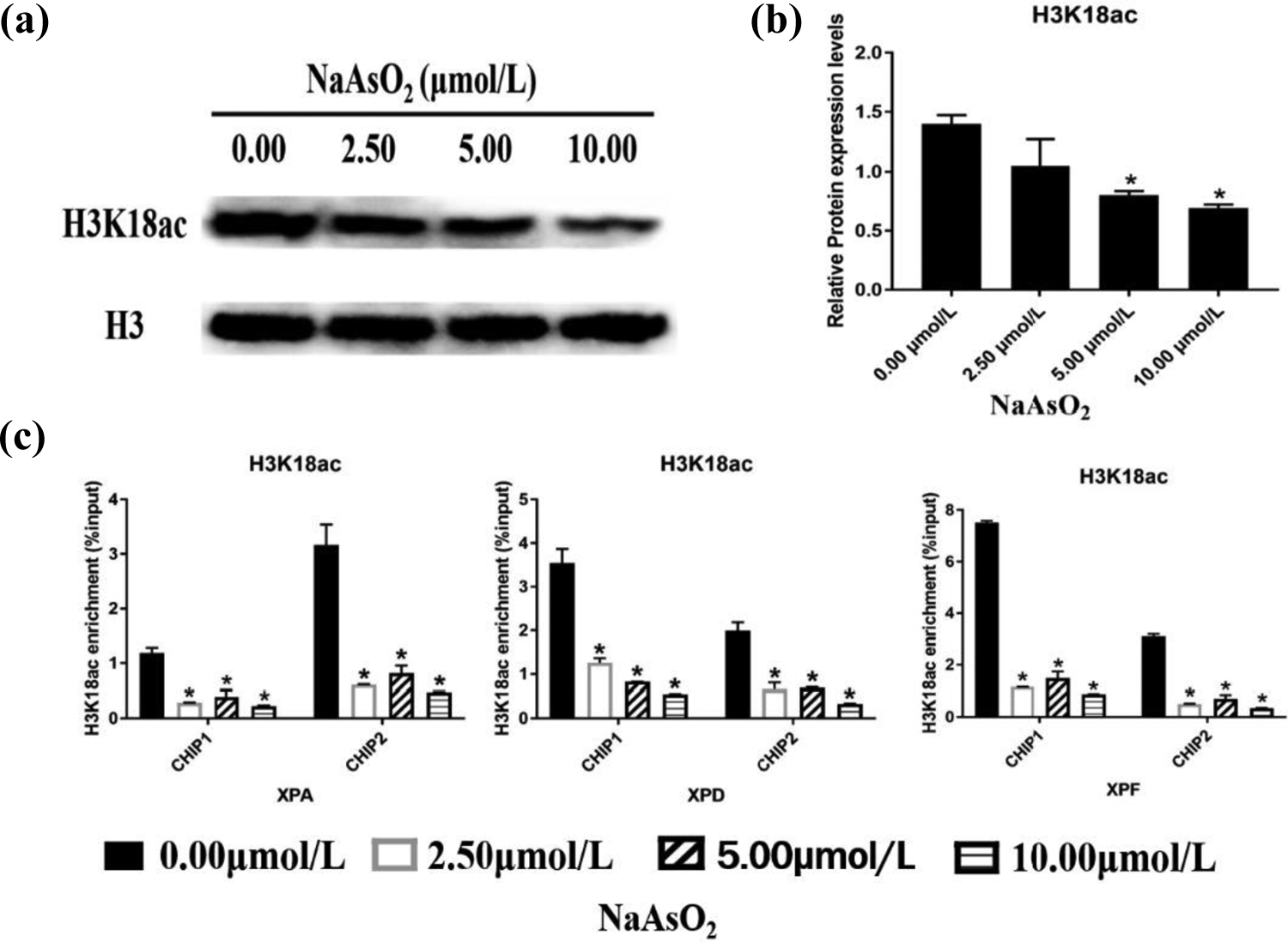
NaAsO2 reduces the level of H3K18 ac modification in the promoter regions of the XPA, XPD, and XPF genes. HaCaT cells were exposed to 0.00, 2.50, 5.00, and 10.00 μmol/L NaAsO2 for 24 h. (a) Western blotting and (b) the relative protein expression levels of H3K18 ac (mean ± SD, n = 3). (c) Enrichment levels of H3K18 ac were analyzed by ChIP using anti-H3K18 ac antibodies and then qPCR was performed. The enrichment levels of modified H3K18 ac in the promoter regions of the XPA, XPD, and XPF genes were expressed as the percentage of chromatin amplification signal intensity following the precipitation of the specific antibody relative to the total input of shear chromatin (mean ± SD, n = 3). IgG was used as negative control. *p < 0.05, compared to the control group. IgG: immunoglobulin G; SD: standard deviation; ChIP: chromatin immunoprecipitation; NaAsO2: sodium arsenite; H3K18 ac: acetylated histone H3K18; XPA: xeroderma pigmentosum A; XPD: xeroderma pigmentosum D; XPF: xeroderma pigmentosum F.
TSA increases the level of H3K18 ac modification in the promoter regions of the XPA, XPD, and XPF genes in arsenic-treated HaCaT cells
TSA is an HDAC inhibitor that increases the degree of histone acetylation by inhibiting the activity of HDACs. 24 Our results showed that the levels of H3K18 ac modification in cells treated with a combination of TSA and NaAsO2 were significantly higher than those in cells from the arsenic group. TSA effectively antagonized histone deacetylation, inhibited the deacetylation of H3K18 ac, and alleviated the NaAsO2-induced decrease in H3K18 acetylation (Figure 3(a) and (b)). The results of ChIP-qPCR analysis showed that, compared to the cells from the arsenic group, H3K18 ac enrichment increased significantly in the gene promoter region in the TSA + NaAsO2 group (Figure 3(c)). This suggests that TSA can increase the H3K18 ac enrichment level in the promoter regions of NER-related genes and, thus, participate in the process of arsenic-induced DNA damage and repair.

TSA increases the H3K18 ac enrichment level in the promoter regions of NER-related genes. The cells were treated with 250 nmol/L TSA for 1 h in advance and then treated with 10 μmol/L NaAsO2 for 24 h. DMSO was used for the control group. (a) Western blotting and (b) the relative protein expression levels of H3K18 ac (mean ± SD, n = 3). (c) ChIP analysis using anti-H3K18 ac antibodies (mean ± SD, n = 3). IgG was used as a negative control. *p < 0.05, compared with the arsenic treatment group. IgG: immunoglobulin G; SD: standard deviation; ChIP: chromatin immunoprecipitation; NER: nucleotide excision repair; NaAsO2: sodium arsenite; H3K18 ac: acetylated histone H3K18.
TSA inhibits the arsenic-induced decrease in the expression of the XPA, XPD, and XPF genes and reduces the DNA damage in HaCaT cells
We found that the level of DNA damage in HaCaT cells in the TSA combined treatment group was slightly higher than that in the control group, but the TailDNA% and OTM values of the TSA combined treatment group cells were significantly lower than those of cells in the arsenic group. These results demonstrate that TSA can alleviate the level of arsenic-induced DNA damage (Figure 4(a) and (b)). The results of the qRT-PCR and Western blotting analyses showed that the mRNA transcript and protein expression levels of the XPA, XPD, and XPF genes in cells from the TSA combined treatment group were significantly higher than those in cells from the arsenic group. Therefore, TSA can effectively antagonize the inhibitory effect of NaAsO2 on the NER activity of the XPA, XPD, and XPF genes and, thus, improve the NER ability of cells (Figure 4(c) to (e)).

TSA inhibits the arsenic-induced decrease in the expression of the XPA, XPD, and XPF genes and reduces DNA damage in HaCaT cells. The cells were treated with 250 nmol/L TSA for 1 h in advance and then treated with 10 μmol/L NaAsO2 for 24 h. DMSO was used for the control group. (a, b) DNA damage was detected by SCGE (Ethidium bromide staining, 200×), and the degree of DNA damage was evaluated based on the OTM and TailDNA% values (mean ± SD, n = 100). (C) The relative expression levels of XPA, XPD, and XPF in arsenic-treated cells following TSA intervention were detected by qRT-PCR (mean ± SD, n = 3). (d, e) Western blotting and the relative protein expression levels of XPA, XPD, and XPF (mean ± SD, n = 3). *p < 0.05, compared to the arsenic treatment group. SCGE: single-cell gel electrophoresis; OTM: olive tail moment; SD: standard deviation; qRT-PCR: quantitative reverse transcription-polymerase chain reaction; NaAsO2: sodium arsenite; XPA: xeroderma pigmentosum A; XPD: xeroderma pigmentosum D; XPF: xeroderma pigmentosum F.
Discussion
Arsenic has been identified as a primary carcinogen by the World Health Organization and the International Agency for Cancer Research, and the carcinogenic mechanism of arsenic is still the focus of attention regarding endemic arsenism. 25,26 Some studies have shown that arsenic-induced genetic damage could be one of the initial factors of arsenic-induced carcinogenesis. 27,28 DNA damage is a highly sensitive molecular biomarker linked to arsenism. 29 Arsenic can also inhibit DNA repair and promote the occurrence of diseases. 30 In our study, to determine the DNA damage induced by arsenic, HaCaT cells were exposed to different concentrations of NaAsO2. The results showed that with increasing arsenic doses, the TailDNA% and OTM values increased significantly, indicating a dose–effect relationship. Thus, we suggest that NaAsO2 can cause notable DNA damage in HaCaT cells.
When DNA damage occurs, cells immediately initiate DNA damage responses to carry out damage repair. Several NER-related genes are involved in DNA damage repair. XPA can recognize DNA damage, stabilize the repaired DNA structure, and recruit downstream repair proteins, playing a key role in the NER process. 31 XPD has lyase activity and is responsible for unlocking the double strand of the damaged DNA. XPF closely binds to the excision repair cross-complementary group 1 (ERCC1) gene, and this complex acts as an endonuclease. Defects or abnormalities in key NER-related genes can lead to a decline in DNA repair ability, increase genomic instability, induce varying degrees of carcinogenesis, and increase cancer susceptibility. 32 In this study, we found that in HaCaT cells exposed to different concentrations of NaAsO2, the expression of NER-related genes was inhibited, and DNA damage repair was abnormal.
A recent study has shown that epigenetics plays an important regulatory role in the process of DNA repair and that histone modifications are involved in DNA replication and DNA damage repair. 33 Histone acetylation can recruit a variety of DNA repair factors and provide a binding platform for these factors. H3K18 ac plays an important role in maintaining genomic integrity and transcriptional regulation. 34,35 Data from a study involving a population subjected to arsenic exposure from drinking water in Bangladesh have shown that arsenic exposure can alter the modification levels of H3K18 ac, H3K27 ac, H3K9me2, H3K27me3, and other histones. 36 It has been shown that arsenic exposure can induce modifications of the histones H3K18 ac, H3K9me2, and H3K36me3 in human lymphocytes. Modification of H3K18 ac may be involved in arsenic-induced oxidative damage by regulating the expression of antioxidant genes. 37 In vivo, it was found that short-term chromium exposure significantly decreased the acetylation level of H3K18 and the expression of 53BP1, which is related to the DNA damage response. At the same time, it was found that the enrichment level of H3K18 ac decreased in the promoter region of the 53BP1 gene. These results show that a decrease in 53BP1 gene expression was related to H3K18 ac. 38 We found that with increasing arsenic concentrations, the level of H3K18 ac modification decreased. The results of ChIP-qPCR analysis using anti-H3K18 ac antibodies showed that the enrichment levels of H3K18 ac decreased gradually in the promoter regions of the XPA, XPD, and XPF genes. We speculate that NaAsO2 can reduce H3K18 ac modifications in the promoter regions of the XPA, XPD, and XPF genes, thus, reducing the mRNA transcript expression of these genes. This demonstrates that H3K18 ac can regulate the transcriptional activation of the XPA, XPD, and XPF genes.
Histone acetylation is a dynamic and reversible modification process. At present, several studies have confirmed that deacetylase inhibitors can change the state of histone acetylation and selectively regulate the transcriptional expression of related genes. 39,40 It has been found that TSA can reduce the degree of DNA damage in pig oocytes. 41 In our study, the mRNA transcript and protein expression levels of NER-related genes increased significantly in arsenic-treated cells that were subjected to TSA intervention, and it was found that TSA inhibited arsenic-induced DNA damage in HaCaT cells. The results of ChIP-qPCR analysis suggested that TSA inhibited the deacetylation of H3K18 in the promoter regions of NER-related genes and increased the H3K18 acetylation level, thus, reducing the affinity between histones and DNA and promoting the transcriptional expression of NER-related genes. Therefore, our intervention study further demonstrated H3K18 acetylation-mediated enhancement of the transcription of the XPA, XPD, and XPF genes.
In summary, in this study, we found that arsenic decreased the expression of NER-related genes, inhibited H3K18 ac modification, reduced H3K18 ac enrichment in the promoter regions of the XPA, XPD, and XPF genes, and increased DNA damage. TSA can effectively alleviate arsenic-induced DNA damage by altering the level of H3K18 ac modification; this provides a new scientific basis for understanding the mechanism underlying arsenic-induced DNA damage.
Supplemental material
Supplementary_materials - Role of H3K18ac-regulated nucleotide excision repair-related genes in arsenic-induced DNA damage and repair of HaCaT cells
Supplementary_materials for Role of H3K18ac-regulated nucleotide excision repair-related genes in arsenic-induced DNA damage and repair of HaCaT cells by AL Zhang, L Chen, L Ma, XJ Ding, SF Tang, AH Zhang and J Li in Human & Experimental Toxicology
Footnotes
Author contribution
The authors AL Z and L C contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of China (project approval numbers: 81430077 and 81360411).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.