
Abstract
Cobalt is a ferromagnetic metal with extensive industrial and biological applications. To assess the toxic effects of, and mechanisms involved in cobalt chloride (CoCl2)-induced cardio-renal dysfunctions. Male Wistar rats were exposed orally, daily through drinking water to 0 ppm (control), 150 ppm, 300 ppm, and 600 ppm of CoCl2, respectively. Following exposure, results revealed significant (p < 0.05) rise in markers of oxidative stress, but decreased activities of catalase, glutathione peroxidase, glutathione-S-transferase, and reduced glutathione content in cardiac and renal tissues. There were significant increases in systolic, diastolic, and mean arterial blood pressure at the 300- and 600-ppm level of CoCl2-exposed rats relative to the control. Prolongation of QT and QTc intervals was observed in CoCl2 alone treated rats. Also, there were significant increases in the heart rates, and reduction in P wave, and PR duration of rats administered CoCl2. Histopathology of the kidney revealed peritubular and periglomerular inflammation, focal glomerular necrosis following CoCl2 exposure. Further, cyclooxygenase-2 and B-cell associated protein X expressions were upregulated in the cardiac and renal tissues of CoCl2-exposed rats relative to the control. Combining all, results from this study implicated oxidative stress, inflammation, and apoptosis as pathologic mechanisms in CoCl2-induced hypertension and cardiovascular complications of rats.
Introduction
Metals, such as cobalt, are natural constituents of all ecosystems and are potent inducers of severe dysregulation of biological systems in man and animals. As a component of vitamin B12, the coenzyme for methyl transferase, required in deoxyribonucleic acid (DNA) biosynthesis, cobalt as is an essential trace element to mammalian life. 1,2 Exposures to high levels of soluble cobalt salts are toxic, with reported median lethal dose ranging from 150 mg kg−1 to 500 mg kg−1 in mammals. 3 In biological systems, cobalt (II) complexes produce oxygen radicals which cause severe deleterious effects on the cardiovascular, hematological, neurological, endocrine, and reproductive systems. 4 –6 Further, Cobalt has erythropoietic action which is attributable to its ability to mimic the pathological response to hypoxia in mammals. 7 Consequently, cobalt salts are used for the treatment of certain types of anemia and as a blood doping agent in humans and race horses. 8 In another documentation, the increased use of cobalt, particularly, in orthopedic surgery and in the manufacture of artificial organs, 9 has raised significant concern about the metal oxide of cobalt (cobalt chloride (CoCl2)) causing cytotoxic, genotoxic, and immunological effects on the recipients of such medical interventions. 10 For instance, cobalt released by metal-on-metal bearing surface and metallic junctions of hip implants have been associated with high incidence of inflammatory pseudo tumor, aseptic lymphocytic vasculitis, and metallosis. 11,12 Although, cobalt is well known to induce hypoxia by inhibiting proline hydroxylase in biological systems, definitive mechanisms for the different toxicities on various organ system are still under intensive investigation. Therefore, this study was designed to investigate the effects of CoCl2 on the cardiovascular and renal systems, while exploring the induction of oxidative stress, inflammation, and apoptosis as probable mechanisms of the CoCl2-induced cardiorenal dysfunctions in the two physiologically related biological systems.
Materials and methods
Chemicals
1,2 Dichloro 4 nitrobenzene, thiobarbituric acid (TBA), trichloro acetic acid (TCA), CoCl2, sodium hydroxide, xylenol orange, potassium hydroxide, reduced glutathione (GSH), O dianisidine, and hydrogen peroxide (H2O2) were purchased from Sigma-Aldrich (St Louis, Missouri, USA). Normal goat serum, biotinylated antibody, and horseradish peroxidase (HRP) system were purchased from KPL, Inc. (Gaithersburg, Maryland, USA). Cyclooxygenase-2 (COX-2) and B-cell associated protein X (Bax) antibodies were purchased from Bioss Inc. (Woburn, Massachusetts, USA) while 3,3′-diaminobenzidine (DAB) tablets were purchased from AMRESCO LLC. (Solon City, Cuyahoga County, Ohio, USA). All other chemicals used were of analytical grade and obtained from British Drug Houses (Poole, Dorset, UK). All other chemicals were of analytical grade.
Experimental animals and design
Forty male rats of the Wistar strain weighing 145–150 g obtained from the Experimental Animal Unit of the Faculty of Veterinary Medicine, University of Ibadan, Nigeria, were used for this study. The rats were randomly divided into four groups of 10 animals each: group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2) orally, daily through drinking water for 7 days. The dosage was chosen based on our previous studies. 13 The rats were kept in wire mesh cages under controlled light cycle (12-h light/12-h dark) and fed with commercial rat chow ad libitum and liberally supplied with water. All the animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” (National Institute of Health, 1996).
Blood pressure measurement
Twenty-four hours after the last administration of CoCl2, blood pressure parameters, including systolic blood pressure (SBP), diastolic blood pressure (DBP), and mean arterial blood pressure (MABP) were determined noninvasively in conscious animals by tail plethysmography using an automated blood pressure monitor (CODA S1, Kent Scientific Corporation, Litchfield city, Connecticut, USA). The average of at least nine readings, taken in the quiescent state, following acclimatization, was recorded per animal.
Electrocardiography
Standard lead II electrocardiogram (ECG) was recorded in rats using a seven-lead ECG machine (EDAN VE-1010, Shanghai, China). The machine was calibrated at 20 mm mV−1 paper speed and 50 mm s−1 paper speed. From the ECG, parameters such as heart rate (HR), P-wave duration, PR-interval, QRS duration, R-amplitude, QT segment, and Bazett’s correction of the QT interval were determined. The rats were anaesthetized with xylazine/ketamine (v/v) 0.1 mL/100 g of rats and administered intramuscularly.
Blood sample collection and plasma preparation
An approximately 3 mL of blood was collected by retro-orbital venous puncture using heparinized capillary tubes into plain bottles and allowed to clot. The clotted blood was then centrifuged at 4000 revolutions per minute (r min−1) for 10 min. Clear serum was separated with Pasteur pipette into another plain tube and then stored at 4°C until needed. The animals were killed by cervical dislocation after blood collection.
Preparation of cardiac and renal homogenates
The organs excised were rinsed and homogenized using 50-mM Tris-hydrochloride (HCl) buffer (pH 7.4) containing 1.15% potassium chloride (KCl). The homogenates were subjected to cold centrifugation at 4°C using a speed of 10,000 r min−1 for 15 min. The post mitochondrial fractions (PMFs) obtained from cardiac and renal homogenates were used for biochemical assays.
Biochemical analysis
Protein concentration was determined by the Biuret method as described by Gornal et al. 14 using bovine serum albumin as standard in heart and kidney tissues. Advanced oxidation protein product (AOPP) levels were determined according to the method of Kayali et al. 15 Briefly, 0.4 mL of cardiac and renal PMFs was treated with 0.8-mL phosphate buffer (0.1 M; pH 7.4). After 2 min, 0.1 mL of 1.16-M potassium iodide was added to the tube followed by 0.2 mL of acetic acid. The absorbance of the reaction mixture was immediately recorded at 340 nm. The concentration of AOPP for each sample was calculated using the extinction coefficient of 261 cm−1 mM−1 and the results were expressed as micromoles per milligram of protein. H2O2 generation was determined by the method described by Wolff. 16 To 2.5 mL of 0.1-M potassium phosphate buffer (pH 7.4), 250 μL of ammonium ferrous sulfate, 100 μL of sorbitol, 100 μL of xynelol orange, 25 μL of sulfuric acid, and 50 μL of sample were added. The mixture was mixed thoroughly by vortexing and a light pink color of the reaction mixture was observed. The reaction mixture was subsequently incubated at room temperature for 30 min. The absorbance was read at 560 nm using distilled water as blank. The H2O2 generated was extrapolated from H2O2 standard curve. Nitric oxide concentrations of PMFs of both the cardiac and renal tissues were measured spectrophotometrically at 548 nm according to the method of Fiddler. 17 The malondialdehyde (MDA) content as an index of lipid peroxidation was quantified in the PMFs of cardiac and renal tissue according to the method of Varshney and Kale. 18 To 1.6 mL of Tris-KCl, 0.5 mL of 30% TCA, 0.4 mL of sample, and 0.5 mL of 0.75% TBA prepared in 0.2-M HCl were added. The reaction mixture was incubated in the water bath at 80°C for 45 min, cooled on ice, and centrifuged at 4,000 r min−1 for 15 min. The absorbance was measured against a blank of distilled water at 532 nm. Lipid peroxidation was calculated with a molar extinction coefficient of 1.56 × 105 M cm−1. The superoxide dismutase (SOD) assay was carried out by the method of Misra and Fridovich, 19 with slight modification from Oyagbemi et al. 20 Briefly, 100 mg of epinephrine was dissolved in 100-mL distilled water and acidified with 0.5-mL concentrated hydrochloric acid. Thirty microliters of PMF were added to 2.5-mL 0.05-M carbonate buffer (pH 10.2) followed by the addition of 300 mL of 0.3-mM adrenaline. The increase in absorbance at 480 nm was monitored every 30 s for 150 s. The one unit of SOD activity was given as the amount of SOD necessary to cause 50% inhibition of the oxidation of adrenaline to adrenochrome. GSH was estimated by the method of Jollow et al. 21 Briefly, 0.5 mL of 4% sulfosalicylic acid (precipitating agent) was added to 0.5 mL of sample and centrifuged at 4000 r min−1 for 5 min. To 0.5 mL of the resulting supernatant, 4.5 mL of Ellman’s reagent (0.04 g of 5, 51- Dithiobis (2- nitrobenzoic acid) (DTNB) in 100 mL of 0.1-M phosphate buffer, pH 7.4) was added. The absorbance was read at 412 nm against distilled water as blank. Catalase (CAT) activity was determined according to the method of Sinha. 22 Briefly, 1 mL portion from the reaction mixture (2 mL of H2O2 solution, 2.5 mL of 0.01-M potassium phosphate buffer (pH 7.0), and 1 mL of properly diluted sample) was blown into 1-mL dichromate/acetic acid solution by a gentle swirl at room temperature at 60 s interval for 3 min into four sets of tubes. The mixture was incubated in the water bath at 100°C for 10 min. The absorbance was read at 570 nm using distilled water as blank. One unit of CAT activity represents the amount of enzyme required to decompose 1 µmol of H2O2/min. Glutathione peroxidase (GPx) activity was also measured according to Beutler. 23 The reaction mixtures contain 0.5 mL of potassium phosphate buffer (pH 7.4), 0.1 mL of sodium azide, 0.2 mL of GSH solution, 0.1 mL of H2O2, 0.5 mL of sample, and 0.6 mL of distilled water. The mixture was incubated in the water bath at 37°C for 5 min and 0.5 mL of TCA was added and centrifuged at 4,000 r min−1 for 5 min. A volume of 1 mL of the supernatant was taken and added to 2 mL of dipotassium phosphate and 1 mL of Ellman’s reagent. The absorbance was read at 412 nm using distilled water as blank. Glutathione-S-transferase (GST) was estimated by the method of Habig et al. 24 using 1-chloro-2,4-dinitrobenzene as substrate. Sulfhydryl (thiol) content was determined as described by Ellman 25 ; 196 μL of potassium phosphate buffer (pH 7.4) was plated in the 96-welled microtiter plate after which 10 μL of sample was added and to 6 μL of DTNB. The reaction mixture was incubated for 30 min at room temperature in a dark room. The assays were done in triplicates and read with BioTek (Houston, Texas, USA) ELx800 Plate reader at 405 nm.
Histopathology
Small pieces of kidney and heart tissues were fixed in 10% formalin, embedded in paraffin wax, and sections of 5–6 mm in thickness were made and thereafter stained with hematoxylin and eosin for histopathological examination according to the methods described by Ellman 25 and Drury et al. 26 Thereafter, the sections were examined with light microscopy.
Immunohistochemical staining for COX-2 and Bax expressions
The immunohistochemistry was described as reported by Oyagbemi et al. 27 To determine the expression of COX-2 and Bax in the heart and kidney, fixed tissues were embedded in paraffin and sectioned at a thickness of 5 μm. The sections were subsequently deparaffinized in xylene and rehydrated with graded alcohol. Antigen retrieval was carried out by immersing the slides in 10-mM citrate buffer at 95–100°C for 25 min with subsequent peroxidase quenching in 3% H2O2/methanol solution. The tissue sections were blocked in goat serum followed by an overnight incubation at 4°C in the goat anti-COX-2 and rabbit anti-Bax primary antibodies. Detection of bound antibody was carried out using biotinylated (goat anti-rabbit, 2.0 µg/mL) secondary antibody and subsequently, streptavidin peroxidase (HRP-streptavidin) according to the manufacturer’s protocol (HistoMark®, KPL, Gaithersburg, Maryland, USA). Reaction product was enhanced with DAB (Amresco®, Solon city, Cuyahoga County, Ohio, USA) for 30 s to a minute and counterstained with high-definition hematoxylin (Enzo®, Farmingdale, NY-USA), with subsequent dehydration in ethanol. The sections were subsequently dehydrated in ethanol and cleared in xylene. The slides were covered with coverslips and sealed with resinous solution. The immunoreactive positive expression of COX-2 and Bax intensive regions was viewed starting from low magnification on each slice then with 400× magnifications using a photo microscope (Olympus, Shinjuku, Tokyo, Japan) and a digital camera (Toupcam®, Touptek Photonics, Zhejiang, China).
Statistical analysis
Data obtained were analyzed with one-way analysis of variance with Dunnett’s posttest at a 95% confidence limit. All values are expressed as mean ± standard deviation. The test of significance between two groups was estimated by Student’s t-test.
Results
The effect of CoCl2 exposure on blood pressure parameters
The SBP, DBP, and MABP of CoCl2 exposed rats increased significantly (p < 0.05) when compared to the control at 300 and 600 ppm of CoCl2 (Table 1).
Blood pressure parameters of rats treated with CoCl2.a

BP: blood pressure; CoCl2: cobalt chloride.
a Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2).
b Significant (p < 0.05) increase compared with control (0-ppm CoCl2). Values are presented as mean ± standard deviation (n = 10). Systolic BP (mmHg), diastolic BP (mmHg), and mean arterial BP (mmHg).
The effect of CoCl2 exposure on electrocardiographic changes
The administration of CoCl2 led to significant (p < 0.05) increases in HRs, QRS, QT, and QTc intervals in comparison with the control (Table 2). However, P wave, R wave, and PR interval declined significantly in CoCl2-treated rats relative to the control (Table 2).
Electrocardiographic changes of rats treated with CoCl2.a

HR: heart rate; CoCl2: cobalt chloride.
a Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2).
b Significant (p < 0.05) increase compared with control (0-ppm CoCl2). Values are presented as mean ± standard deviation (n = 10).
c Significant (p < 0.05) decrease compared with control (0-ppm CoCl2). Values are presented as mean ± standard deviation (n = 10).
The effect of CoCl2 exposure on markers of oxidative stress and antioxidant defense system
The activities of cardiac and renal GST and GPx and CAT decreased (p < 0.05) significantly in rats exposed to different levels of CoCl2 compared with the control, with the greatest reduction in activity occurring in rats exposed to 600-ppm CoCl2 (Table 3).
Cardio and renal antioxidant enzyme status in rats treated with CoCl2.a

GSH: reduced glutathione; CoCl2: cobalt chloride; GPx: glutathione peroxidase (units/mg protein); GST: glutathione-S-transferase (mmole 1-chloro-2, 4 dinitrobenzene-GSH complex formed/min/mg protein); SOD: superoxide dismutase (units/mg protein); CAT: catalase (mmoleH2O2 consumed/min/mg protein); H2O2: hydrogen peroxide.
a Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2).
b Significant (p < 0.05) difference compared with control (0-ppm CoCl2). Values are presented as mean ± standard deviation (n = 10).
In a dose-dependent manner, exposure of rats to CoCl2 led to significant (p < 0.05) increases in the markers of oxidative stress (H2O2 generation and MDA contents) in both the cardiac and renal tissues of rats at different doses compared with the control group (Figures 1 and 2). Protein thiol (PSH) content increased significantly in the kidney tissues of CoCl2-exposed rats at 600 ppm (Figure 3). Also, a significant (p < 0.05) reduction was observed in the GSH level of both the cardiac and renal tissues of CoCl2-exposed rats compared to the control group (Figure 4), but the level of AOPP increased significantly in all exposed rats relative to the control (Figure 5).

Effect of CoCl2 on H2O2 generation in the hearts and kidneys of exposed rats. Values are presented as mean ± standard deviation. aStatistically significant when groups B, C, and D are compared with group A. Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2). CoCl2: cobalt chloride; H2O2: hydrogen peroxide.

Effect of lead acetate on malondialdehyde levels in the hearts and kidneys of exposed rats. Values are presented as mean ± standard deviation. aStatistically significant when groups B, C, and D are compared with group A. Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2). CoCl2: cobalt chloride.

Effect of cobalt chloride on sulfhydryl thiol content in the hearts and kidneys of exposed rats. Values are presented as mean ± standard deviation. aStatistically significant when groups B, C, and D are compared with group A. Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2). CoCl2: cobalt chloride.
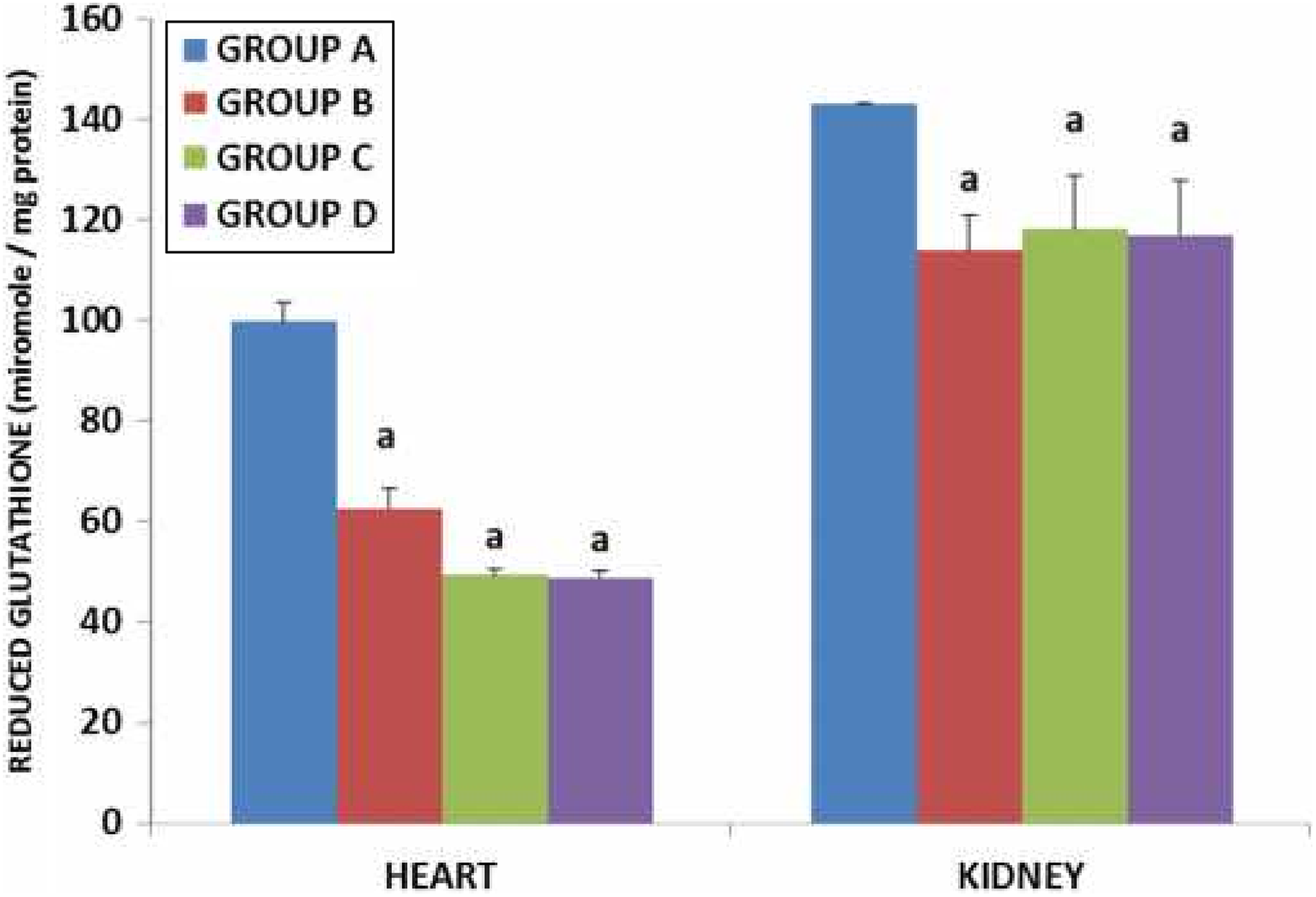
Effect of CoCl2 on reduced glutathione in the hearts and kidneys of exposed rats. Values are presented as mean ± standard deviation. aStatistically significant when groups B, C, and D are compared with group A. Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2). CoCl2: cobalt chloride.

Effect of CoCl2 on advanced oxidation protein product in the hearts and kidneys of exposed rats. Values are presented as mean ± standard deviation. aStatistically significant when groups B, C, and D are compared with group A. Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2). CoCl2: cobalt chloride.
Histopathology
Hisopathological observations of kidney tissues revealed peritubular and periglomerular inflammation in rats exposed to 150-ppm CoCl2, focal area of glomerulonecrosis in rats exposed to 300 ppm, and fusion of the glomeruli with the Bowman’s capsule in rats exposed to 600-ppm CoCl2 (Figure 6). However, visible lesions were absent in the cardiac tissues of rats (Figure 7).

Histological observation of the kidney. (a) Control: no visible lesion; (b) 150-ppm CoCl2: plate shows peritubular and periglomerular inflammation (black arrow); (c) 300-ppm CoCl2: plate shows peritubular and periglomerular inflammation (black arrow) and a focal area of glomerulonecrosis (green arrow); and (d) 600-ppm CoCl2: plate shows the fusion of the glomeruli with the Bowman’s capsule (black arrow). Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2). Magnification ×100 (H & E). CoCl2: cobalt chloride; H & E: Haematoxylin and Eosin.

Histological observation of the kidney. (a) Control: no visible lesion; (b) 150-ppm CoCl2: no visible lesion; (c) 300-ppm CoCl2: no visible lesion; and (d) 600-ppm CoCl2: no visible lesion (black arrow). Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2). Magnification ×100 (H & E). CoCl2: cobalt chloride.
Immunohistochemistry of cardiac and renal COX-2 and Bax
Immunohistochemistry revealed higher immunopositive expressions of cardiac and renal Bax and COX-2 in rats administered 150-, 300-, and 600-ppm CoCl2 compared to the control rats in a dose-dependent manner (Figures 8 and 9).

The effect of CoCl2 intoxication of cardiac Bax expressions. Group A (control) shows lower expressions of Bax in the hepatic tissues. Group B (150 mg L−1 of CoCl2) shows higher expressions of Bax. Group C (30 mg L−1 of CoCl2) higher expressions of Bax than groups A and B cells, whereas the rats in group D (600 mg L−1 of CoCl2) show the expressions of Bax higher than that of group C (black arrows). The slides were counterstained with high-definition hematoxylin and viewed ×400 objectives (magnification ×100). Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2). CoCl2: cobalt chloride.

The effect of CoCl2 intoxication of cardiac COX-2 expressions. Group A (control) shows the lower expressions of COX-2 in the hepatic tissues. Group B (150 mg L−1 of CoCl2) shows the higher expressions of COX-2. Group C (300 mg L−1 of CoCl2) shows the higher expressions of COX-2 than groups A and B cells, whereas the rats in group D (600 mg L−1 of CoCl2) show the expressions of COX-2 higher than that of group C (black arrows). The slides were counterstained with high-definition hematoxylin and viewed ×400 objectives (magnification ×100). Group A (control, received water), group B (received 150-ppm CoCl2), group C (300-ppm CoCl2), and group D (600-ppm CoCl2). COX-2: cyclooxygenase-2; CoCl2: cobalt chloride.
Discussion
The interrelationships between oxidative stress, inflammation, and apoptosis in toxicant-induced physiological derangements have been reported by various authors. 28,29 The hypoxic response induced by CoCl2 is reportedly associated with increase production of reactive oxygen species (ROS) leading to cellular damage and apoptosis in biological systems. 30,31 CoCl2 functions as an oxidative stress-inducing agent because Co (II) reacts directly with H2O2 in a Fenton-type reaction to produce ROS which in excess can affect cell survival through process that irreversibly alters the DNA. 32,33 However, the generated ROS could be reduced by intracellular antioxidant enzymes such as GPx and CAT as well as some nonenzymatic antioxidant molecules like glutathione and vitamin E, respectively. 34 CoCl2-induced cytotoxicity coupled with the generation of ROS has also been described. 35 In this study, CoCl2 precipitated significant reductions in the activities of the enzymatic antioxidants (GPx, GST, and CAT) and nonenzymatic intracellular antioxidant (GSH) together with increase in the level of the markers of oxidative stress (MDA, H2O2, AOPP, and sulphydryl thiol content) in a dose-dependent manner. These observations suggest a potent induction of oxidative stress and hence corroborate earlier reports describing oxidative stress as a mechanism of CoCl2-induced toxicity in various organs. 36 –40
The observed elevated MDA contents led to enhanced lipid peroxidation products and this might be due to the production of superoxide anion, peroxyl, and hydroxyl radicals. 41 The increased peroxidation of membrane lipids is one principal consequence of oxidative damage produced by cobalt. 42 Furthermore, the toxicity of CoCl2 has been severally reported to aggravate the generation of hydroxyl radicals (·OH) from H2O2, causing significant reduction in GSH level and increase lipid peroxidation products such as MDA and other thiobarbituric reaction substances. 43 Therefore, the observed significant reduction in the activities of GPx, GST, and CAT may have resulted from a staggering production of ROS or an inactivation and/or decreased production of the antioxidant enzymes. More so, that the protective mechanisms described for antioxidants against toxicities associated with acute and chronic exposures to metals via different pathways include trapping of free radicals, chelating metal ions and preventing the reaction with ROS, and maintaining metal ions in a redox state. 44 The GST is an enzyme that is particularly important as a catalyst in the metabolism of reactive substances of exogenous or endogenous origin. 45 Furthermore, the inhibitory effect of CoCl2 on antioxidant enzymes has been reported elsewhere. 46,47 Also, in this study, the sulfhydryl thiol content and AOPP increased significantly in renal tissues of rats exposed to 600 ppm of CoCl2. Thiol containing antioxidants such as N-acetyl-L-cysteine have been reported to mitigate drug and toxicant-induced oxidative stress-mediated injuries and prevented the depletion of hepatic and plasma GSH level by scavenging intracellular ROS. 48 Overall, CoCl2 potentially precipitated oxidative stress and free radical generation with concomitant organ damage.
From our laboratory, we have described the negative impact of CoCl2 on blood pressure parameters and the use of synthetic and natural antioxidants for the amelioration of CoCl2. 49,50 Oxidative stress has been suggested as a pathogenic factor and therapeutic target in early stages of essential hypertension, with the SBP and DBP correlating positively with reduction in the total antioxidant capacity of plasma in hypertensive subjects. 51 Hence, enzymatic and nonenzymatic antioxidants can mitigate the development of oxidative stress and associated dysregulation of biological systems through several mechanistic processes including a direct scavenging activity on ROS/reactive nitrogen species (RNS) or inhibition of ROS/RNS formation. 52 In the present study, the observed significant increases in the blood pressure parameters (SBP, DBP, and MABP) of rats administered CoCl2 alone when compared to control were consistent with the induction of oxidative stress which is a major cause of endothelial dysfunction, primarily, through the reduction in nitric oxide bioavailability. 53,54 In addition, the HR was increased, P wave and PR interval duration of rats administered CoCl2 was shortened. Thus, the electrocardiographic abnormalities may suggest an activation of a physiological protective mechanism in response to decreased cardiac contractility occasioned by the hypoxia/ischemia normally accompanying prolonged CoCl2 exposure. Interestingly, observation of elecrocardiographic disturbances is absent in animals administered single intravenous dose of CoCl2, 55 but sub-chronic coadministration of CoCl2 with ethanol and sugar is reported to cause severe cardiac abnormalities in rats. 56 Moreover, prolonged exposure to CoCl2 has been reported to cause significant reductions in the activities of cardiac enzymes such as manganese superoxide dismutase, succinate cytochrome c oxidase, nicotinamide adenine dinucleotide (reduced form) (NADH) cytochrome c reductase, and severe decreased ability of the cardiac mitochondria to utilize metabolic substrate for adenosine triphosphate (ATP) production. 57
Similarly, free radicals such as superoxide anion (·O2 −) inactivate nitric oxide, which is an endogenous vasodilator, and cause a reduction in the level of tetrahydrobiopterin, an essential cofactor for the synthesis of nitric oxide (NO) in biological systems. 58 ROS-induced reduction in the bioavailability of NO causes severe impairments to the intrinsic vasodilatory properties especially in the arteriolar vascular beds, increases systemic vascular resistance, and elevates blood pressure beyond the normal physiological range. 59 Under normal conditions, SOD dismutates ·O2 − to H2O2, which is subsequently converted to water and oxygen by CAT and GPx. However, oxidative stress inducing chemicals, such as COCl2, exacerbate the production of ROS and RNS beyond the coping capacity of the antioxidant defense systems with consequent deleterious effects. For instance, an elevated H2O2 level, as observed in this study, may cause a reduction in the antioxidant activity of CAT via an inactivation of the enzyme or a downregulation of its gene expression. 60 The enzymatic antioxidants such as SOD and GPx have been reported to decrease significantly in untreated hypertensive patients. 61
Furthermore, oxidative stress has been reported to be actively involved in COX-2-mediated elevation of blood pressure. 62 From our study, CoCl2 induced a dose-dependent increase in COX-2 expressions in the kidney tissues of rats. COX-2 is expressed transiently by a wide spectrum of growth factors and pro-inflammatory stimuli under certain pathophysiological conditions. 63 Furthermore, diets deficient in salt, inhibition of angiotensin converting enzyme, administration of diuretics, and/or experimental renovascular hypertension are reported to increase COX-2 expression in the kidney, by inducing a high renin state. 64 The increase expression of COX-2 in the kidney of rats exposed to different levels of CoCl2, as observed in this study, suggests an activation of the renin angiotensin system and may be attributable to the severe hypertensive states of the toxicant exposed rats.
Also, in this study, CoCl2 caused a dose-dependent increase in Bax expressions in the heart and kidney tissues of rats. Bax is a proapoptotic member of the Bcl-2 family, 65 whereas COX-2 is the inducible prostaglandin endoperoxide synthase, and COX-2 is involved in the synthesis of eicosanoids (prostaglandins, prostacyclin, thromboxane, and leukotrienes). 66 Apoptosis is reported to cause increase in cardiac pathologies, 67 through different pathways including cytokine/Fas receptor-driven pathway, mitochondrial-driven pathway, and endoplasmic reticulum/Ca2+-driven pathway. 68 The mitochondrial-mediated pathway, which is the best characterized of the three apoptotic pathways, plays a pivotal role in the apoptotic process and includes the Bax protein and the involvement of Bcl-2 has been documented in the regulation of mitochondria permeability. 69 In the present study, the dose-dependent increased expressions of the proapoptotic Bax protein observed in the cardiac and renal tissues of rats exposed to CoCl2 suggest an involvement of the mitochondrial mediated pathway in CoCl2-induced cardio-renal toxicity. It is probable that CoCl2 stimulated the translocation of Bax to the mitochondria and triggered a conformational change in structure that allows Bax-Bax oligomerization and insertion of Bax into the outer mitochondrial membrane with subsequent rapid release of apoptogenic proteins such as cytochrome C. 70 Moreover, the hypoxic interior milieu associated with CoCl2 intoxication stimulates the translocation of cytosolic heat shock protein 60 to the plasma membrane, releases Bax, and possibly speeds up the oligomerization process, mitochondrial pore formation, and apoptosis. 71,72 Therefore, the increased Bax protein expressions in the cardiac and renal tissues of rats exposed to different doses of CoCl2 suggest a disruption of the protective pathway involved in the confinement of Bax to the cytosol and aggravation of the apoptotic pathway by CoCl2. Observations in this study are similar to earlier reports of increase caspases-3 and -9, and Bax protein expression in CoCl2-induced apoptosis. 73,74 Further, the marker of oxidative stress AOPP, which increased significantly in the CoCl2 exposed rats, has been implicated in the pathogenic processes leading to apoptosis via the motion of nicotinamide adenine dinucleotide phosphate (reduced form) (NAPDH) oxidase-dependent podocyte depletion in the glomerulus by a p53-Bax apoptotic pathway both in vivo and in vitro. 75 AOPPs are dityrosine-containing protein products, formed during oxidative stress, 76 that have been found to play important roles as renal pathogenic mediators of apoptosis in glomerular cells and are reported to increase in chronic renal insufficiency, diabetes, and hypertension. 77
Altogether, CoCl2 administration caused hypertension, precipitated electrocardiographic changes, increased markers of oxidative stress, and declined antioxidant defense system. Also, markers of inflammation and apoptosis were dose-dependently increased following exposure to CoCl2. Combining all, caution must be taken in the use of CoCl2 for recreational and therapeutic purposes due to the aforementioned toxicity which could portend public health risk.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.