
Abstract
Previous studies showed that paraquat (PQ) caused the apoptosis of dopaminergic neurons by inducing the generation of oxygen radical. The purpose of this study is to explore PQ-induced microglial inflammatory response and its underlying molecular mechanisms. The murine microglia BV2 cell line was used. After stimulation with PQ and lipopolysaccharides (positive control), the concentrations of tumor necrosis factor-α (TNF-α), interleukin 1β (IL-1β), and interleukin 6 (IL-6) in the culture supernatant and mRNA expression of TNF-α and IL-1β were determined by ELISA and quantitative real-time Polymerase Chain Reaction (PCR), respectively. The protein expression of heat shock protein 60 (HSP60) and toll-like receptor 4 (TLR4), along with the mRNA expression of transcription factors of nuclear factor κB-p65 (NF-κB-p65) and activated protein 1 (AP1, c-fos, and c-jun dimer) were evaluated with western blot and quantitative real-time PCR, respectively. The results showed that PQ activated microglia, which was characterized by increasing the generation and upregulated mRNA expression of pro-inflammatory cytokines, TNF-α, IL-1β, and IL-6. In addition, PQ significantly enhanced the expressions of HSP60 and TLR4 proteins in BV2 cells, as well as NF-κB-p65, c-fos, and c-jun mRNA. These findings suggest that PQ can activate microglia and enhance the expression and secretion of pro-inflammatory cytokines in a HSP60/TLR4 signaling, leading to the inflammatory response.
Introduction
Microglia is the primary immune cells of the central nervous system (CNS) and they are highly similar to peripheral macrophages. After acute or chronic exposure to some chemicals or stressors, microglia is activated. Activated microglia release a large number of pro-inflammatory cytokines to induce an inflammatory response, which disturbs the normal neurobiological function and ultimately contributes to progression and severity of the disease. 1,2 The massive generation of microglia-mediated inflammatory mediators can lead to neuronal loss; therefore, inflammation is considered as a contributor to the pathogenesis of neurodegenerative disorders. 3 The principal mechanism that initiates microglial response is involved in the toll-like receptors (TLRs), primarily expressed on microglia as a complement of pattern recognition receptors. 4 TLRs canbe activated by some molecules as ligands, such as lipopolysaccharides (LPS) and heat shock protein 60 (HSP60), to induce inflammation cascade. 5 –7 As an essential member of TLRs family, the pathophysiological role of TLR4 in the CNS received more attention. In vivo and in vitro studies demonstrated that some endogenous and exogenous chemicals, such as Aβ and morphine, activated TLR4 signaling cascade and downstream nuclear factor κB (NF-κB) signaling pathway in microglia induces a pro-inflammatory response by releasing some inflammatory cytokines, which includes tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6) and interleukin 1β (IL-1β), and so on, which finally result in neuronal degeneration and acceleration of the pathological progression of neurodegeneration. 1,5,8
Paraquat (N,N′-dimethyl-4,4′-bipyridinium dichloride, PQ), as a herbicide, is commonly implicated with the elevated incidence of Parkinson’s disease (PD). Epidemiological studies showed an increased risk of developing PD in human populations living in areas where PQ exposure is likely to occur and among workers lacking appropriate protective equipment. 9 –11 Laboratory studies involving repeated PQ exposure in animal models showed the decreased or altered locomotor activity in conjunction with the pathological features of PD, including significant loss of dopaminergic neurons, degeneration of nigrostriatal dopamine system, and aggregate formation in the substantianigra pars compacta containing α-synuclein and formation of Lewy bodies. 12 –14 Oxidative stress and the metabolic processes of PQ-inducing excitotoxicity, α-synuclein aggregate formation, autophagy, alteration of dopamine catabolism, and inactivation of tyrosine hydroxylase are positioned as the mechanisms of PQ-involved in developing PD. 15 The neuronal death in neurodegenerative diseases can activate microglia through TLR4 signaling, causing microglial cells to secrete additional inflammatory factors, which further leads to neuronal damage. Given these connections, we hypothesized that PQ could separately induce the inflammatory response of microglia by enhancing HSP60 expression and the activation of TLR4 signaling pathways. Therefore, in the present study, BV2 cells were used as a microglial model to explore whether PQ was able to induce a microglial inflammatory response by activating HSP60/TLR4 signaling at the molecular level.
Materials and methods
Chemicals
PQ (paraquat dichloride hydrate, 99% of purity) and LPS were obtained from Sigma Co., Ltd (Missouri, USA). The stock solutions of PQ (10 mM) and LPS (1 mg/mL) were prepared by dissolving them in distilled water, and they were kept at 4°C away from light. Before use, they were diluted into the corresponding application solution with culture medium.
Cells culture and treatment
Murinemicroglial line BV2 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) at 37°C under an atmosphere of 5% CO2. All cells culture products were purchased from Gibco (California, USA), except that FBS was from NQBB in Australia. BV2 cells were treated with PQ at the levels of 0 μM, 20 μM, 40 μM, and 80 μM and LPS at 1 μg/mL (positive control), respectively, for 6 h, 12 h, and 24 h.
Measurement of pro-inflammatory cytokines
After indicated treatments, the culture supernatant was collected and the levels of TNF-α, IL-6, and IL-1β were measured using ELISA kits of TNF-α (Joyee, Shanghai, China), IL-6 and IL-1β (Boster, Wuhan, China), respectively, according to the manufacturer’s instructions. The experiments were repeated for three times.
Western blot
Protein was extracted using radio immunoprecipitation assay (RIPA) Lysis Buffer (Beyotime, Shanghai, China) and was measured with bicinchoninic acid (BCA) protein quantification kit (Beyotime, Shanghai, China). Total protein samples were separated by 12% SDS-PAGE and transferred to PVDF membrane. After blocking at a room temperature for 2 h with 5% non-fat dry milk, the blocked membranes were incubated with primary antibodies against TLR4 (Wanlei, Shenyang, China), HSP60 (Immunoway, Missouri, USA), and β-actin (Wanlei, Shenyang, China) overnight at 4°C. Then, the membranes were incubated with the secondary antibodies (Wanlei, Shenyang, China) at a room temperature for 1 h. Finally, the bands were visualized using an enhanced chemiluminescence reagent, and they were analyzed using the image software and normalized to β-actin.
Quantitative real-time Polymerase Chain Reaction (PCR)
After treatment, cells were washed with phosphate buffer saline (PBS) and total RNA was isolated with TRIzol reagent (Invitrogen, California, USA) following the manufacturer’s instructions. The One Step SYBR®PrimeScriptTM PLUS RT-PCR Kit (TaKaRa, Dalian, China) was used for the quantitative real-time PCR (qRT-PCR) analysis. The qRT-PCR was performed at 95°C for 15 s, followed by 40 cycles at 95°C for 5 s and 60°C for 34 s at ABI 7500 real-time PCR detection system. Primers were designed using Primer 5 system and synthesized by Takara (Dalian, China). GAPDH was used as the endogenous control. The primer sequences were as follows: for NF-κB-p65 (NM_009045.4) sense, 5′-GTGTCTTGGTGGTATCTGTGC-3′ and NF-κB-p65 anti-sense, 5′-GATCTGTTTCCCCTCATCTTT-3′; c-fos (NM_010234.2) sense, 5′-CTGTCACCGTGGGGATAAAGT-3′, and c-fos anti-sense, 5′-GACCATGATGTTCTCGGGTTT-3′; c-jun (NM_010591.2) sense, 5′-TAGAACGGTCCGTCACTTCAC-3′, and c-jun anti-sense, 5′-CGCACGCTCCTAAACAAACTT-3′; TNF-α (NM_001278601.1) sense, 5′-TGAAGGGAATGGGTGTTCAT-3′, and TNF-α anti-sense, 5′-GAGTTGGACCCTGAGCCATA-3′; IL-1β (NM_008361.4) sense, 5′-CAGGCAGGCAGTATCACTCA-3′ and IL-1β anti-sense, 5′-TGTCCTCATCCTGGAAGGTC-3′; GAPDH (NM_001289726.1) sense, 5′-AAGAAGGTGGTGAAGCAGG-3′ and GAPDH anti-sense, 5′-GAAGGTGGAAGAGTGGGAGT-3′. The data were obtained by normalizing target gene cycle threshold (Ct) values with corresponding glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Ct, and then analyzed with 2−ΔΔCt method. All experiments were repeated three times.
Statistical analysis
All data were expressed as mean ± SD of three independent experiments. The SPSS 17.0 software was used for statistical analysis. Two group comparisons were performed with student t-test and multiple group comparisons were analyzed with least significant difference (LSD) of one-way analysis of variance (ANOVA). A significant difference was considered for p < 0.05.
Results
PQ induced the morphological changes of microglia
Normally, microglia in the “resting” state appear in branch shape, showing clear body and protuberances. However, once it is stimulated, microglia have a transformation from “resting” state to “activated” state, which appear in amoebic shape, showing enlarged body, shortened or disappeared protuberances. As anticipated, after PQ exposure, morphological alteration of BV2 cells was observed and their protuberances gradually became shortened and bodies became round (Figure 1), which were more pronounced with increasing dosage and duration of PQ exposure. These changes suggested that PQ induces the activation of microglia.
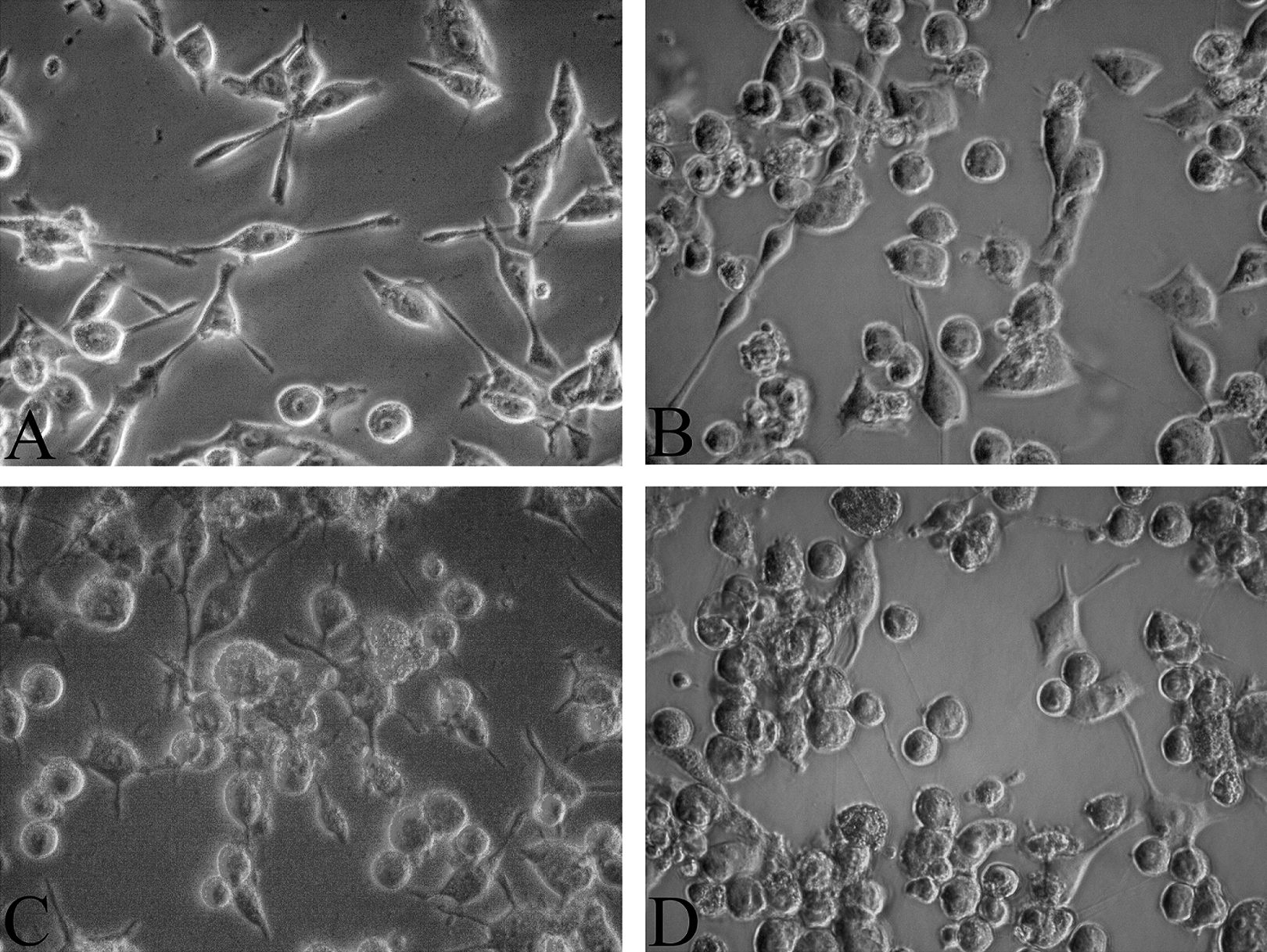
PQ induced the morphological alteration of BV2 cells. BV2 cells appear in branch shape in resting state. After the stimulation with LPS and PQ, BV2 cells are transformed to activated state and appear in amoebic shape, which protuberances became shortened and bodies became round. (a) control group; (b) LPS group; (c) 40 µM PQ group; (d) 80 µM PQ group. LPS: lipopolysaccharides; PQ: paraquat.
PQ increased concentration and mRNA expression of pro-inflammatory cytokines
Compared with the normal control group, the levels of TNF-α, IL-1β and IL-6 in the culture supernatant increased significantly in time- and dose-dependent manner in PQ exposure groups (p < 0.05), as shown in Figure 2. However, the increment of TNF-α, IL-1β, and IL-6 concentrations in PQ groups were weaker than that in LPS positive control group (p < 0.05). To further explore whether PQ increases the expression of pro-inflammatory substances at a molecular level, TNF-α and IL-1β mRNA expression were examined in BV2 cells exposed to PQ. qRT-PCR results (Table 1) showed that the expression of TNF-α and IL-1β increased significantly in PQ group and LPS positive control group for 6, 12, and 24 h (p < 0.05), compared with the negative control group. For 6, 12, and 24 h, PQ (40 µM) increased mRNA expression of TNF-α by 31.7, 63.3, and 42.6%, and mRNA expression of IL-1β by 31.3, 37.5, and 34.4%, respectively.

The concentrations of pro-inflammatory cytokines in the culture supernatant of BV2 cells. PQ exposure activated microglia to secrete (a) TNF-α, (b) IL-1β, and (c) IL-6. *Compared with the control group, p < 0.05. PQ: paraquat; TNF-α: tumor necrosis factor-α; IL-6: interleukin 6; IL-1β: interleukin 1β.
Relative mRNA expression of TNF-α and IL-1β in BV2 cells (X ± S, n = 3).

TNF-α: tumor necrosis factor-α; IL-6: interleukin 6; IL-1β: interleukin 1β; LPS: lipopolysaccharides; PQ: paraquat.
a p < 0.05, compared to control group at the same duration.
b p < 0.05, compared to LPS group at the same duration.
PQ increased protein expression of HSP60 and TLR4
Since HSP60 can mediate the activation of microglia through TLR4 pathway, 16 we hypothesized that HSP60 expression in activated microglia was increased to further aggravate the immune response. As shown in Figure 3, the protein level of HSP60 increased significantly in response to PQ and LPS treatment at all point in time (p < 0.05), compared to the control. Also, we detected the TLR4 protein expression in BV2 cells. Figure 3 showed that TLR4 expression increased time-dependently after PQ treatment, similar to that exposed to LPS. These results suggest that PQ enhances the protein expression of HSP60 and TLR4 in microglia and thus launches the TLR4 cascades.

PQ increased the expression of HSP60 and TLR4 in BV2 cells. (a) The protein expression of HSP60 and TLR4 were detected using western blot. (b) Quantification of HSP60 and TLR4 expression was expressed as the ratio (in percentage) of β-actin. *Compared to the control group, p < 0.05. PQ: paraquat; HSP60: heat shock protein 60; TLR4: toll-like receptor 4.
PQ induced mRNA expression of NF-κB-p65 and AP1
Induction of TLR4 can initiate the downstream signaling cascades; therefore, it can trigger the activation of NF-κB and activated protein 1 (AP1). AP1 is a homologous or heterogenous dimer of c-fos and c-jun. As shown in Figure 4, the results from qRT-PCR showed that mRNA expression levels of NF-κB-p65, c-jun, and c-fos increased in BV2 cells treated by PQ, indicating that downstream NF-κB and AP1 signaling is activated after PQ exposure, which then induces the increased expression of pro-inflammatory cytokines and further aggravates the inflammatory response in CNS.

Effects of PQ on mRNA expression of NF-κB-p65 and AP1 in BV2 cells. The mRNA expression of (a) NF-κB-p65, (b) c-fos, and (c) c-jun was up-regulated in BV2 cells after PQ treatment at different time. *Compared to the control group, p < 0.05. PQ: paraquat; NF-κB-p65: nuclear factor κB-p65; AP1: activated protein 1.
Discussion
Stressed, injured, and dying cells release the signals into their local environment, where these signals send the warning of abnormal cellular function to the organism and activate resident innate immune cells, mainly microglia in the CNS. Activated microglia release a large number of pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, and so on, to induce an inflammatory response. 2 Similar result to these studies were observed in such a way that PQ induced the transformation of microglia from resting to an activated state and increased the expression of TNF-α and IL-1β mRNA, as well as the release of TNF-α, IL-1β, and IL-6 out of the cells. These findings indicate that PQ induces a microglia-mediated inflammatory response. Due to the facts of long-term or chronic inflammation contributing to the progression and severity of the disease in the CNS, we consider that PQ-induced sustained inflammatory response is able to lead to the CNS damage and might play a role in the pathogenesis of neurodegenerative disorders, which would be focused on in our future research. However, the mechanism of PQ-induced inflammatory response of microglia is the central point in the present study.
Some molecules released from injured or dying eukaryotic cells have been identified to activate innate immunity. Heat shock proteins, constitutively expressed in most cells under cellular stress conditions, nutrient deprivation, or mechanical damage, are considered as endogenous dangerous signals to the immune system. 17 HSP60 induces maturation and activation of innate immune cells and plays immune regulation effects in case of various stress or injury. 18,19 Studies have demonstrated that the enhanced expression of HSP60 was induced in microglia stimulated by IL-1β, then HSP60 activated TLR4 pathway in a myeloid differentiation factor 88 (Myd88)-dependent pathway and its downstream signaling of the inflammatory process, which result in the increase of production of pro-inflammatory factors and the alert of occurrence of CNS injury. 7,16,20 Thus, HSP60 is considered as an endogenous activator of TLR4 and it also plays an important role in increasing the intensity of inflammation with its continuous production and serves as a general danger signal for CNS injury. In our study, significantly increased expression of HSP60 and TLR4 was observed after PQ stimulation, suggesting that PQ promotes the enhanced expression of HSP60 in the microglia, and then HSP60 further binds to and upregulates TLR4 expression. Therefore, we speculate that HSP60 likely modulates the intracellular signaling of PQ-induced inflammation, which requires further investigation in future studies.
Activated TLR4 recruits MyD88 through interaction with TLR domains and initiate intracellular signaling cascades, leading to the activation of downstream transcription factors of NF-κB and AP1, which are the transcription regulators of a number of cytokines that is responsible for cellular immune response, as well as the generation of pro-inflammatory mediators. 21 It is reported that CNS inflammatory response can be mediated by TLR4/NF-κB signaling pathway in vitro and in vivo. 22 NF-κB is sequestered in the cytoplasm in an inactive form. Once stimulated, NF-κB-p65, a subunit of NF-κB, is shifted into the nucleus, where activated NF-κB-p65 binds to the promoters of its target genes and regulates the expression of the gene of pro-inflammatory mediators through the activation of the phosphoinositide 3-kinase and mitogen-activated protein kinase cascades. 23,24 So NF-κB is considered as a major regulator of inflammatory responses and its activation causes nuclear translocation of NF-κB-p65 and subsequent expression of pro-inflammatory mediators. 25,26 The inhibition of NF-κB can reverse the expression of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, which can be induced by some chemicals or stimulation. 25 In the present study, the upregulation expression of NF-κB-p65 mRNA was observed, indicating that PQ exposure activates NF-κB signaling. AP1, composed of homodimers or heterodimers of c-fos and c-jun proteins that are crucial for cell adaptation to many environmental changes. 27 The c-fos and c-jun gene are expressed constitutively in certain tissues and are induced to express rapidly and transiently in response to multiple stimuli and combine to form AP1 in the form of leucine zipper structure with transcriptional activation activity. 28 After the combination of AP1 with DNA, the expression of downstream target genes, such as pro-inflammatory cytokines gene, are modulated to participate in physiological processes. As anticipated, an increase in the mRNA levels of c-jun and c-fos was observed in PQ-stimulated BV2 cells, indicating that PQ exposure enhances the transcriptional activity of AP1, and then results in the increased expression of pro-inflammatory cytokines. Currently, it is unclear whether the effects of PQ on the activation of NF-κB and AP1 were directly modulated by suppression of TRL4 signaling, which requires further investigation.
Collectively, PQ can activate microglia and induce inflammatory responses. PQ-induced microglial inflammation involves in HSP60 as an endogenous signal that further relays inflammation via TLR4 inside the microglia. The suppression of neuroinflammation by inhibiting microglial activation and the generation of pro-inflammatory cytokines could be a worthwhile strategy for attenuating PQ neurotoxicity in the future.
Footnotes
Author contribution
Yuan-yuan Sun and Jing Zheng contributed equally.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Special Fund for Scientific and Technological Innovation in Harbin City of China (2017RAXXJ070) and Outstanding Youth Training Foundation in Harbin Medical University of China (no grant number).