
Abstract
Recent studies have reported the potential of pyrano-pyridine compounds in inhibiting cell growth and apoptosis induction in cancer cells. Here, we investigated the effect of new pyrano-pyridine derivatives on proliferation, oxidative damages, and apoptosis in K562 leukemia cells. Among different tested compounds, we found 8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5, 6, 7, 8-tetrahydro-6-phenethyl-4H-pyrano-[3,2-c]pyridine-3-carbonitrile (4-CP.P) as the most effective compound with IC50 value of 20 μM. Gel electrophoresis, fluorescence microscopy, and flow cytometry analyses indicated the apoptosis induction ability of 4-CP.P in K562 cells. Further analyses revealed that 4-CP.P induces significant increase in cellular reactive oxygen species production, lipid peroxidation, protein oxidation, and total thiol depletion. Interestingly, while 4-CP.P significantly increased the activity of superoxide dismutase, it reduced the catalase activity in a time-dependent manner. These data propose that 4-CP.P treatment causes free radicals accumulation that ultimately leads to oxidative stress condition and apoptosis induction. Therefore, we report the 4-CP.P as a novel, potent compound as a chemotherapeutic agent in cancer treatment.
Introduction
The most common approaches in cancer treatment are surgery, radiation, and chemotherapy. In previous decades, chemotherapy was the most conventional method for human leukemia treatment. Indeed, valid evidence obtained in recent years shows that many chemotherapeutic agents exert their antitumor potential through triggering the mechanism of apoptosis. 1 Apoptosis is identified as an active programmed cell death in order to remove harmful cells and prevent uncontrolled cell proliferation. 2 There are several morphological hallmarks in this process including cell shrinkage, pyknosis, chromatin condensation, plasma membrane blebbing, and DNA fragmentation. 3 In mammalian cells, factors that trigger apoptotic pathways include radiation, toxins, hypoxia, hyperthermia, viral infections, and free radicals such as reactive oxygen species (ROS) and reactive nitrogen species. 4 ROS, including superoxide (O2 −), hydrogen peroxide (H2O2), and the hydroxyl free radical (HO−), are continuously generated from many cellular sources at low concentrations under physiological conditions. ROS have been shown to influence cell cycle progression, apoptosis, and growth factor signaling in different types of cells. 5,6 In contrast, antioxidant defense system has been proven to neutralize the immeasurable levels of free radicals. These defense mechanisms contain enzymes such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX). In general, several small molecules such as glutathione (GSH), vitamins E and C, β carotene, and selenium have potential of eliminating free radicals. 7 Overproduction of ROS and/or absence of suitable compensatory response from antioxidant complex lead to a wider state known as oxidative stress. 8 In this condition, high ROS levels are responsible for the oxidative damages of biomolecules including DNA, proteins, sugars, and lipids. 7 Accordingly, increased oxidative stress is related to various diseases including a wide range of tumor types. 9 It should be noted that there is a complex association between cellular stress and cancer. It has been proposed that the increased levels of ROS and oxidative stress can induce cancer. However, the higher production of ROS by malignant cells compared to their normal cellular counterparts suggests the anticancer potential of these molecules. 10 Therefore, in cancer cells, ROS is a double-edged sword. At low levels, it is required for cancer cell survival due to the cell cycle progression, as well as regulation of chronic inflammation. On the other hand, excessive levels can repress tumor progression through effect on cell cycle inhibitor proteins and/or induction of cell death mechanism such as apoptosis and necrosis. 11,12 In fact, many cytotoxic drugs used in cancer treatment kill cancer cells by amplifying ROS stress. 13 There are numerous synthetic compounds that have been investigated and examined for cancer treatment. However, many of these compounds are not suitable for therapeutic purposes due to their toxic, carcinogenic, and mutagenic properties. 14 In recent decades, significant types of heterocyclic compounds were discovered as pyrano-pyridines, which have wide range of biological activities including antitumor, anti-inflammatory, antifungal, antibacterial, and antimalarial. 15 –17 In the present study, we examined the cytotoxic effect of four compounds of pyrano-pyridine family on K562 human chronic myeloid leukemia cell line (Figure 1). 18 After primary assessments, 8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5, 6, 7, 8-tetrahydro-6-phenethyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile (4-CP.P), as an active pyrano-pyridine derivate, was selected for evaluation of the possible effect of oxidative stress on apoptosis induction in K562 leukemia cells.
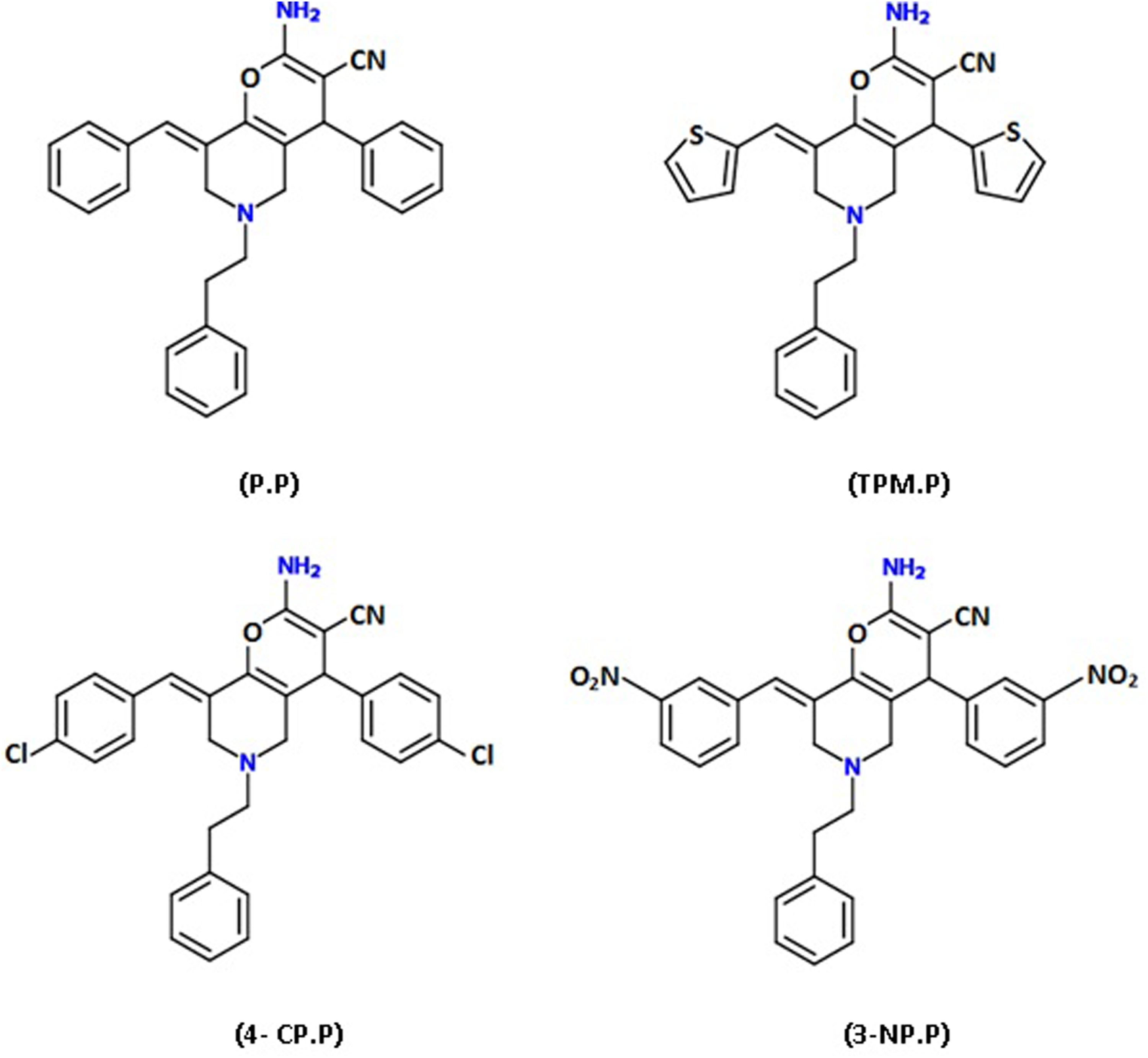
Chemical structure of the investigated pyrano-pyridine derivatives.
Materials and methods
Materials
The cell culture medium (RPMI 1640), fetal bovine serum (FBS), and penicillin–streptomycin were purchased from Gibco BRL (Life technologies, Paisley, Scotland). The cell culture plates were obtained from SPL (Korea). Ethidium bromide (EtBr), acridine orange (AO), proteinase K, 3-(4, 5-dimethylthiazol-2-yl) 2, 5-diphenyl tetrazolium bromide (MTT) reagent, 2′, 7′-dichloro fluorescein diacetate (DCFH-DA) reagent, RNase A, and dimethylsulfoxide (DMSO) were obtained from Sigma (Germany). Annexin-V FITC apoptosis kit, propidium iodide (PI), and cell extraction buffer were purchased from Roche (Germany). 2, 4 Dinitrophenylhydrazine (DNPH) and 5,5′ dithiobis-2-nitrobenzoic (DTNB) were purchased from Merck (Germany). The human K562 cell line was obtained from Pasteur Institute (Tehran, Iran).
Preparation of the investigated pyrano-pyridine derivatives
2-Amino-4-aryl-8-[(E)-arylmethylidene]-5, 6, 7, 8-4H pyrano-[3, 2-c]pyridine derivatives were synthesized in peptide chemistry research center, K. N. Toosi University of Technology. Briefly, reaction of 3, 5-bis [(E)-arylmethylidene]-tetrahydro-4 (1H)-pyridinones with malononitril in aqueous media and in the presence of diammonium hydrogen phosphate (10%) or piperidine (10%) leads to pyrano-pyridines at room temperature. The structures of target compounds including 2-amino-8-benzylidene-5, 6, 7, 8-tetrahydro-6-phenethyl-4-phenyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile (P.P), 2-amino-5, 6, 7, 8-tetrahydro-6-phenethyl-4-thiophen-2-yl-8-(thiophen-2-yl)methylene) 4H-pyrano-[3, 2-c]pyridine-3-carbonitrile (TPM.P), 4-CP.P, and 8-(3-nitrobenzylidene)-2-amino-5, 6, 7, 8-tetrahydro-4-(3-nitrophenyl)-6-phenethyl-4H Pyrano-[3, 2-c]pyridine-3-carbonitrile (3-NP.P) were established by IR, 1H-NMR, C-NMR, as well as mass spectrometry and elemental analysis. 18
Cell culture and treatment
The human chronic myelogenous leukemia K562 cells were cultured in RPMI-1640 medium supplemented with FBS (10%, v/v), streptomycin (100 µg/ml), and penicillin (100 U/ml) in CO2 humidified atmosphere at 37°C.
Cell viability assay
MTT was used as an indicator of cell viability as determined by its mitochondrial-dependent reduction to formazone. To examine the effects of the pyrano-pyridine derivatives on the cell proliferation, the K562 cells (1 × 105 cells/well) were seeded into 96 well plates for 24 h prior to treatment. After this period, the cells were treated with different concentrations (20–100 µM) of the compounds for 24–72 h. After washing with phosphate buffered saline (PBS), MTT (0.5 mg/ml) was added to each well 4 h prior to harvesting. Formazone crystals were dissolved using DMSO. Finally, the absorbance was read at 570 nm with a microplate reader (Expert 96, Asys Hitech, New Zealand). The results were reported as survival percent using the following equation

where As is referred to the mean absorbance read for each sample, A 0% and A 100% are the absorbencies read for the DMSO treated (dead) and untreated cells, respectively. Untreated cells and DMSO treated cells were used as negative and positive controls, respectively.
Apoptosis assay
Apoptotic morphological changes
The cells (5 × 105 cell/ml) were seeded in 12 well plates and treated with single dose (at IC50 value) of 4-CP.P for 72 h. Apoptosis was determined morphologically by AO-EtBr staining using fluorescence microscopy. In brief, after washing cells with PBS, 25 µl of the cell suspension was added to 1 ml of prepared solution (100 µg of each dyes/ml PBS) in 1:1 volume ratio and evaluated by fluorescence microscopy (Olympus BX41, Germany). 19 The morphology of untreated and the 4-CP.P treated cells were also studied using an inverted microscope (Zeiss, Germany).
DNA fragmentation assay
The K562 cells treated with IC50 value of 4-CP.P were collected after 72 h, washed with PBS and then incubated in 1 ml of lysis buffer (0.01 M Tris-HCl (pH 8.0), 0.1 M NaCl, 0.025 M Ethylenediaminetetraacetic acid (EDTA) (pH 8.0), and 1% Sodium dodecyl sulfate [SDS]), which contained 10 µg/ml RNase A for 1 h at 37°C, followed by 0.2 mg/ml proteinase K for 2 h at 50°C. After centrifugation at 12,000×g, the supernatant was extracted with phenol/chloroform/isoamyl alcohol (25:24:1) to remove the proteins and lipids. DNA in the supernatant was then precipitated in ethanol overnight at 20°C and pelleted at 15,000×g. The dry sample pellets were resuspended in Tris-HCl-EDTA (TE) (10 mM Tris-HCI, pH 7.9, 1 mM EDTA) buffer and loaded into the 1.5% agarose gel containing EtBr, and then electrophoresed for 2 h. 20
Cell cycle analysis
For cell cycle analysis, the cells (5 × 104 cell/well) were grown in 25-cm2 cell culture flasks 24 h prior to treatment. The treated cells with a single dose of 4-CP.P were collected after 72 h and washed twice with PBS. Then fixed in 70% ethanol and kept at −20°C until analysis. The cells were then stained with 50 μg/ml PI containing 20 μg/ml RNase (DNase free) for 2 h. The percent of calculated cells in the sub-G1, G0/G1, S, and G2/M phases were determined by flow cytometry (BD FACSCalibur flow cytometer, Biosciences, San Jose, CA, USA). 21
Flow cytometric assessment of apoptosis
Annexin-V is a phosphatidylserine-binding protein that interacts strongly and specifically with phosphatidylserine residues and can be used for the detection of apoptosis. 22 In this test, an annexin-V FITC/PI double staining method was used according to the manufacture’s protocol. Briefly, after double washing of treated and untreated cells with PBS, 1 × 106 cells were resuspended in binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Then, 5 µl of annexin-V FITC and 5 µl PI were added. The mixture was incubated for 15 min in dark at room temperature and then measured by flow cytometry (BD FACSCalibur flow cytometer, USA).
Assay of intracellular ROS levels
One of the most widely used methods for the investigation of the intracellular ROS generation involve oxidizable fluorescent dyes, such as acetylated forms of DCFH-DA. This dye is a stable compound that readily diffuses into cells and is hydrolyzed by intracellular esterase to DCFH which is trapped within the cells. Reactive oxygen species (ROS) including H2O2 or low-molecular weight hydroperoxides produced by cells, oxidize DCFH to the highly fluorescent compound, 2′, 7′-dichlorofluorescein (DCF). Thus, the fluorescence intensity is proportional to the amount of peroxide produced by the cells. 23 Herein, after 72 h, K562 cells treated with IC50 value of 4-CP.P were treated with 10 µM DCFH-DA for 30 min. Then, the cells were washed twice with PBS to remove the extracellular compounds, and DCFH-DA fluorescence was detected using flow cytometry (Becton Dickinson, Biosciences, San Jose, CA, USA).
Assays for cellular redox markers
For all enzyme activity assays in the present study, cell lysates were required. After treatment of the cells for various time intervals, the cells were harvested and washed twice with PBS. Then the cells were homogenized by adding lysis buffer (100 µl per 1,000,000 cell) and vortexed every 10 min for half hour and finally, centrifuged in 13,000 r/min for 10 min. The supernatant (cell lysate) was subjected to assay under specific conditions (−80° C). The cellular protein concentration was determined by Bradford assay. 24 SOD and CAT activities were measured by pyrogallol autoxidation 25 and Aebi’s 26 methods, respectively. The measurement of total thiol content was carried out using 5, 5′ dithiobis-2-nitrobenzoic acid (DTNB) by Ellman’s method. 27 Measurement of malondialdehyde (MDA) formation was carried out through its reaction with thiobarbituric acid (TBA). 28 Protein oxidation was measured by Reznick method with modifications. 29
SOD activity measurement
Fifty microliters of the supernatant were added to a 1 ml reaction mixture consisting of 50 mM Tris-Cl buffer and 1 mM EDTA in pH 8.2 (blank). Fifty microliters of pyrogallol (0.2 mM) were added to reaction medium and the absorbance decrease of pyrogallol was monitored at 420 nm for 15 min. Results were expressed as SOD units per mg of soluble cell protein measured according to the Bradford method, using bovine serum albumin as standard. 24
CAT activity measurement
A mixture of 3 ml of Tris buffer with 25 µl of sample was selected as reaction medium (the absorbance at 240 nm was recorded as blank). Twenty-five microliters H2O2 were added to reaction medium, the activity was determined following the disappearance of H2O2 at 240 nm by using an ultraviolet spectrophotometer (T60, PG Instruments Ltd, Leicestershire, UK). Results were expressed as units per mg of soluble protein.
Determination of lipid peroxidation
Among lipids, those containing unsaturated fatty acids are particularly susceptible to interact with free radicals. The most common method used for lipid peroxidation assay is the estimation of aldehydic products particularly MDA by their ability to react with TBA that yields thiobarbituric acid reactive substances (TBARS), which can be easily measured by spectrophotometry. 30 In this assay, after exposure of K562 cells to certain amount of 4-CP.P (IC50 value), the cells were mixed with 0.4 ml of 2.8% trichloroacetic acid (TCA) and 0.6 ml of 0.67% TBA and heated to 95°C for 1 h. After cooling, 1.5 ml of n-butanol was added followed by vigorous shaking. The protein concentration was determined by the Bradford method. 24 The total amount of TBARS to form chromophores absorbing at 532 nm was estimated using a molar absorption coefficient of 1.56 × 105 M−1 cm−1. The results are expressed as nmol TBARS per mg of protein. 28
Determination of protein oxidation
Protein carbonyls (PCO) were measured by using the method of Reznick. 29 Briefly, 1 ml of supernatant was placed in two glass tubes. Then 2 ml of 10 mM DNPH in 2.5 M HCl was added to one of the tubes, while 2 ml HCl (2.5 mM) was added to the other tube. Tubes were incubated for 1 h at room temperature. Samples were vortexed every 15 min. Then 2.5 ml TCA (20%, w/v) was added and the tubes were left on ice for 5 min followed by centrifugation for 5 min to collect the protein precipitates. The pellet was then washed three times with 2 ml ethanol–ethyl acetate (1:1, v/v). The final precipitate was dissolved in 1 ml 6 M guanidine hydrochloride solution and incubated for 10 min at 37°C while mixing. The absorbance of the sample was measured at 370 nm. The carbonyl content was calculated based on the molar extinction coefficient of DNPH (ε = 2.2×104 cm− 1 M− 1) and expressed as nmol/mg protein.
Total thiol (−SH) content
Total −SH groups were measured using DTNB as the Ellman’s reagent, in which it reacts with the SH groups to produce a yellow colored complex which has a peak absorbance at 412 nm. Briefly, 1 mL Tris-EDTA buffer (pH 8.6) was added to 50 μL cell lysate and the absorbance was read at 412 nm against Tris-EDTA buffer alone (A1). Then, 20 μL DTNB reagent (10 mM in methanol) was added to the mixture and after 15 min (stored in room temperature), the sample absorbance was read again (A2). The absorbance of DTNB reagent was also read as the blank (B). Total thiol concentration (mM) was calculated using the following equation
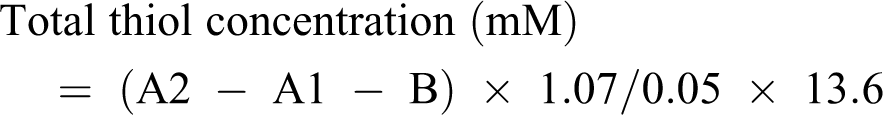
Statistical analyses
All experiments were done in triplicate and the data are presented as mean ± SD. Comparison between groups was made by one-way analysis of variance (ANOVA) followed by a specific post hoc test to analyze the difference.
Results
Cell viability and growth inhibition
To determine the effect of the investigated compounds on the viability of the K562 cells, 1 × 105 cells/ml were treated with various concentrations (10–50 µM) of the compounds for 24, 48 and 72 h. All tested compounds were cytotoxic against K562 cells, in a dose- and time-dependent manner (Table 1). As shown in Table 2, and Figure 2 after 72 h exposure of the cells, the IC50 (concentration that inhibits 50% of cell survival) values of P.P, TPM.P, 4-CP.P, and 3-NP.P were calculated as 60 ± 4.0, 100 ± 3.0, 20 ± 3.0, and 80 ± 5.0 µM, respectively. Based on these data, 4-CP.P was the most effective derivative against K562 cells compared to other compounds. As shown in Table 1 and Figure 1, at 10–50 µM, 4-CP.P reduced viability of the cells by 40–70% after 72 h. Based on these data, we selected the 4-CP.P with IC50 value of 20 µM for further analysis.
Effect of pyrano-pyridine derivatives on the K562 cell viability.a

MTT: 3-(4, 5-dimethylthiazol-2-yl) 2, 5-diphenyl tetrazolium bromide; P.P: 2-amino-8-benzylidene-5, 6, 7, 8-tetrahydro-6-phenethyl-4-phenyl-4H-pyrano-[3, 2-c]Pyridine-3-carbonitrile; TPM.P: 2-amino-5, 6, 7, 8-tetrahydro-6-phenethyl-4-thiophen-2-yl-8-(thiophen-2-yl)methylene) 4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; 4-CP.P: 8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5, 6, 7, 8-tetrahydro-6-phenethyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; 3-NP.P: 8-(3-nitrobenzylidene)-2-amino-5, 6, 7, 8-tetrahydro-4-(3-nitrophenyl)-6-phenethyl-4H-pyrano[3, 2-c]pyridine-3-carbonitrile; SD: standard deviation.
aThe cells were treated with various concentrations (20–100 µM) of the compounds for 24, 48, and 72 h. Cell viability was evaluated by MTT assay. Data were expressed as a percentage of control measured in the absence of the compounds. Each point represents the mean ± SD of three independent experiments (p < 0.05).
The IC50 values of the investigated compound after 24–72 h.
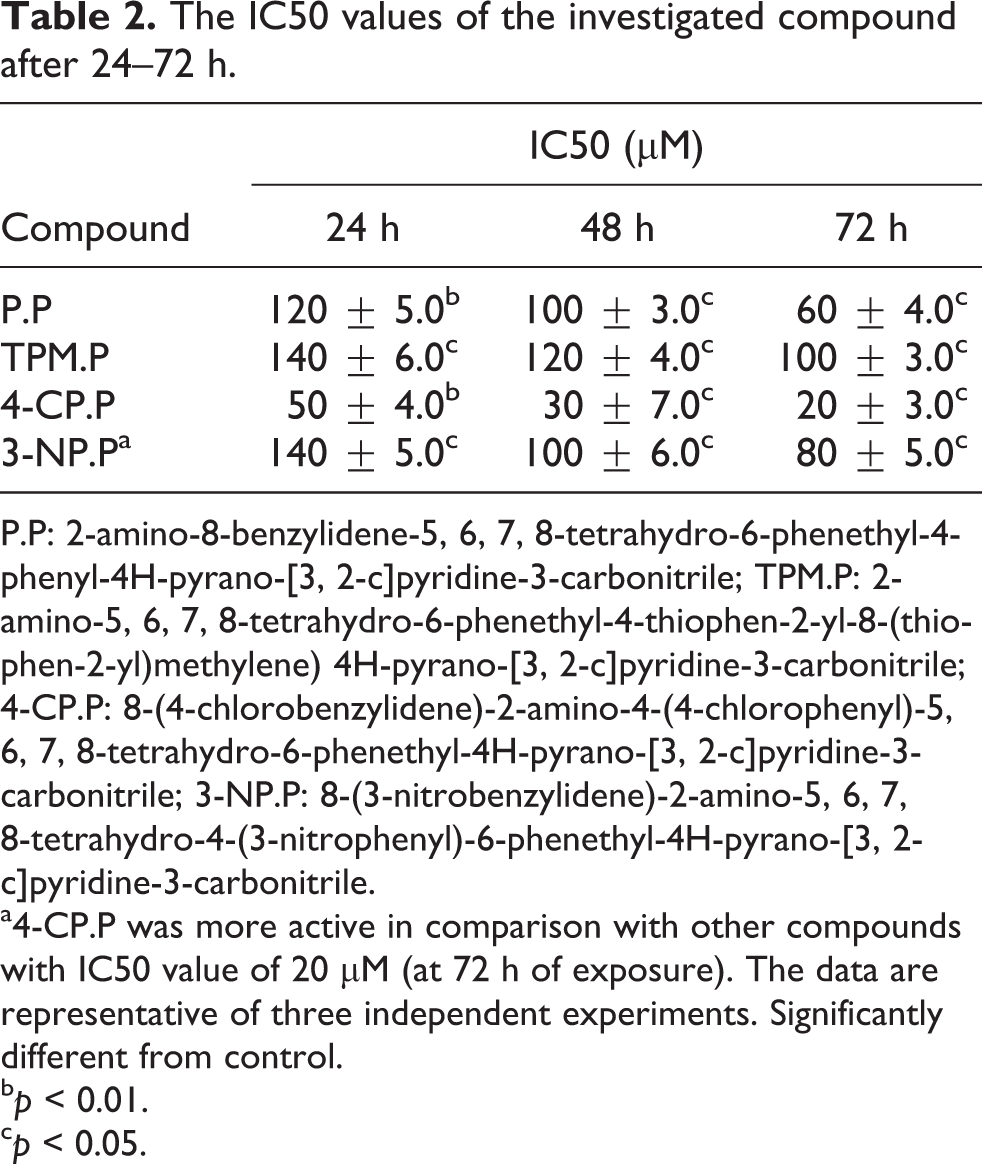
P.P: 2-amino-8-benzylidene-5, 6, 7, 8-tetrahydro-6-phenethyl-4-phenyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; TPM.P: 2-amino-5, 6, 7, 8-tetrahydro-6-phenethyl-4-thiophen-2-yl-8-(thiophen-2-yl)methylene) 4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; 4-CP.P: 8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5, 6, 7, 8-tetrahydro-6-phenethyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; 3-NP.P: 8-(3-nitrobenzylidene)-2-amino-5, 6, 7, 8-tetrahydro-4-(3-nitrophenyl)-6-phenethyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile.
a4-CP.P was more active in comparison with other compounds with IC50 value of 20 µM (at 72 h of exposure). The data are representative of three independent experiments. Significantly different from control.
b p < 0.01.
c p < 0.05.

Viability of the K562 cells upon exposure to the 4-CP.P. The K562 cells were treated with various concentrations (10–50 µM) of the 4-CP.P for 24–72 h. The cell viability was determined by MTT reduction assay. Data are presented as mean ± SEM (n = 3). *p < 0.05; **p < 0.01 versus control. 4-CP.P: 8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5, 6, 7, 8-tetrahydro-6-phenethyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; MTT: 3-(4, 5-dimethylthiazol-2-yl) 2, 5-diphenyl tetrazolium bromide; SEM: standard error of mean.
Induction of apoptosis
Morphological changes of the K562 cells
To evaluate apoptosis, the cells were cultured at a density of 1 × 105 cells/ml and treated with the indicated concentration (IC50 values) of 4-CP.P. The morphological changes were inspected by AO/EtBr dual staining. In this approach, after staining, live cells are green, while yellow and red represent cells that are at the early and late apoptosis stages, respectively (Figure 3(A, b)). In addition, appearance of untreated cells were perfectly round in comparison with treated cells, which showed plasma membrane blebbing and apoptotic bodies after 72 h under light and fluorescence microscope (Figure 3 (A, a and b)). We further confirmed the apoptosis event through analysis of DNA ladders formation (Figure 3 (B, a)).

Investigation of morphological changes, cell cycle analysis, and apoptosis induction in K562 cells treated with 4-CP.P. (A, a) Light microscope image of the K562 cells. Untreated cells have a round shape indicating live cells. While, treated cells after 72 h have a condensed and fragmented form. (A, b) Fluorescence images of the K562 cells stained with AO/EtBr. 4-CP.P induced membrane blebbing and nuclei fragmentation marked with arrows. (B, a) Fragmentation pattern of extracted DNA upon treatment with the compound for 72 h as compared to the controls. C: control, M: marker. (B, b) The cell cycle distribution was analyzed using flow cytometry. Our findings showed that the 4-CP.P induces sub-G1 cell cycle arrest in K562 cells after 72 h. (C) For evaluation of apoptosis, the cells were treated with 20 µM 4-CP.P for 72 h, then were harvested and apoptosis was studied by annexin-V/PI double staining as mentioned in materials and methods. The results suggested apoptosis induction in the cells after 72 h. 4-CP.P: 8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5, 6, 7, 8-tetrahydro-6-phenethyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; AO/EtBr: acridine orange/ethidium bromide; PI: propidium iodide.
Apoptosis assay by flow cytometry
To support the previous results demonstrating the apoptosis induction by 4-CP.P, we studied the cell cycle distribution pattern of the cells treated with the compound. DNA content analysis of treated K562 cells after PI staining showed the appearance of sub G1 peak (representing population of apoptotic cells) after 72 h (Figure 3 (B, b)). As exhibited in Figure 3 (B, b), the control cells were distributed among sub G1, G0/G1, S, and G2/M phases by 7.1%, 42.2%, 31.3%, and 19.5%, respectively. After treatment of the cells with 20 µM 4-CP.P for 72 h, a significant increase (9.69 fold) in the sub G1 cell population was observed compared to the control cells. To quantitate the apoptosis assessment, we investigated the redistribution of the plasma membrane of the cells (as hallmark of apoptosis) to visible phosphatidylserine after double staining with annexin-V/PI. Consistent with described data, apoptosis was mostly observed after 72 h of exposure to 4-CP.P (20 µM). The result of flow cytometric analysis showed that the percent of late apoptotic and/or necrotic cells (upper right quadrant) compared to the control cells increased after 72 h. Based on Figure 3(C), shifts from early apoptosis (lower right quadrant) to late apoptosis or necrosis (upper right quadrant) are clearly evident in dot plots (data not shown).
Measurement of ROS generation
Studies have shown that chemotherapeutic agents often induce apoptosis in cancer cells through increasing ROS generation or decreasing ROS scavenging capacity. 31 Therefore, we analyzed the oxidative stress condition in the cells treated with the 4-CP.P. To examine the effect of 4-CP.P on intracellular ROS production, we used DCFH-DA staining. As shown in Figure 4, the generation of ROS in K562 cells increased time dependently after 24–72 h of exposure to 4-CP.P (20 µM). So that, the ROS level was significantly increased (43.01%) compared to the control group (6.28%) after 72 h of K562 cells treatment with 4-CP.P (Figure 4).

The effects of 4-CP.P on intracellular ROS production. K562 cells were treated with 4-CP.P (20 µM) for 24, 48, and 72 h. Then, cells were incubated with DCFH-DA and the fluorescence intensity of 10,000 cells was analyzed using a flow cytometry. The results demonstrated that 4-CP.P was potent at creating a state of oxidative stress induced apoptotic cell death. 4-CP.P: 8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5, 6, 7, 8-tetrahydro-6-phenethyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; ROS: reactive oxygen species; DCFH-DA: 2′, 7′-dichlorofluorescein diacetate.
The effect of 4-CP.P on stress oxidative parameters in K562 cells
In order to detect the effect of 4-CP.P on the cellular redox status in K562 cells, antioxidant defense system capabilities, lipid peroxidation, and protein oxidation were evaluated. Superoxide anion radicals (O2 ·−) as major ROS generated in mitochondria are converted to H2O2 by SOD. Subsequently, CAT is one of the several mechanisms that regulate the H2O2 level. 32 As demonstrated in Figure 5 (A), after 24, 48, and 72 h treatment of the K562 cells with 20 µM 4-CP.P, the SOD activity was significantly elevated to 26.07, 54.88, and 64.41 U/mg Pr, respectively compared to the control cells (11.27 U/mg Pr). In contrast, the CAT activity (Figure 5 (B)) was reduced to 13.63 U/mg Pr after 3 days in treated cells versus 35.51 U/mg Pr in untreated group. As shown in Table 3, total thiol content reduced time dependently after 24–72 h. After 72 h incubation of the cells with 20 µM 4-CP.P, the amount of total thiol reduced to 11.5 ± 2.12 µmol/mg Pr that was about 2.5 fold lower than untreated cells (29 ± 1 µmol/mg Pr). Furthermore, the significantly increased TBARS levels were the lipid peroxidation indicator in K562 cells treated with IC50 value (20 µM) of the compound (Table 3). The most widely studied marker of ROS-mediated protein oxidation is accumulation of PCO groups, which are formed by direct oxidation of protein side chains or indirect lipid peroxidation that can be introduced into proteins. 33 Our results also showed that, 4-CP.P caused a marked increase in the PCO content (0.76 ± 0.28 nmol/mg protein) after 72 h of exposure to the compound versus control cells (0.33 ± 0.01 nmol/mg protein) (Table 3). These data suggest that 4-CP.P triggers apoptosis in K562 cells through induction of oxidative damage.
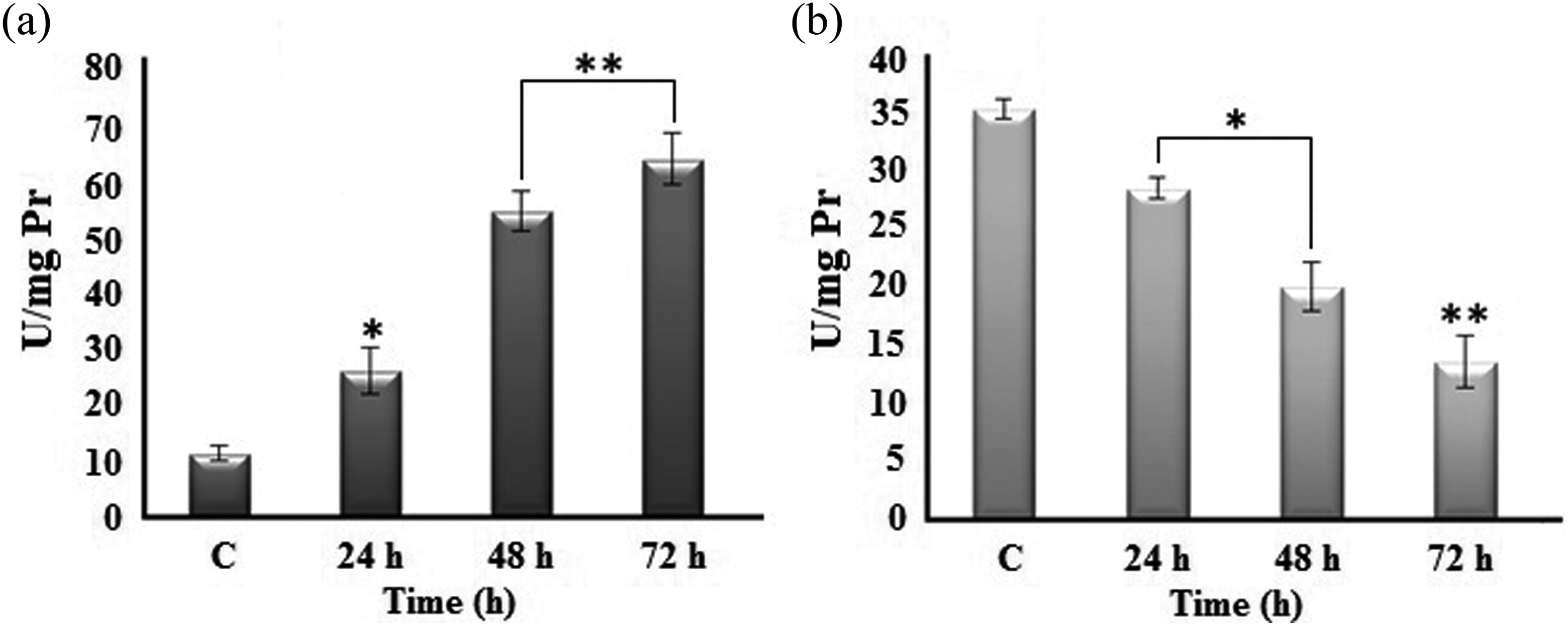
Effect of 4-CP.P on SOD (a) and CAT (b) activities in K562 cells. Each value represents the mean ± SD (n = 3). Statistical analysis was performed by one-way ANOVA, followed by the post hoc test. *p < 0.05; **p < 0.01. 4-CP.P: 8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5, 6, 7, 8-tetrahydro-6-phenethyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; SOD: superoxide dismutase; CAT: catalase; ANOVA: analysis of variance; SD: standard deviation.
Effects of 4-CP.P on PCO formation, lipid peroxidation (TBARS) and total thiol content in K562 cells.a

4-CP.P: 8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5, 6, 7, 8-tetrahydro-6-phenethyl-4H-pyrano-[3, 2-c]pyridine-3-carbonitrile; PCO: protein carbonyl; TBARS: thiobarbituric acid reactive substances; ANOVA: analysis of variance; SD: standard deviation.
aEach value represents the mean ± SD (n = 3). Statistical analysis was performed by one-way ANOVA, followed by the post hoc test.
b p < 0.05.
c p < 0.01.
Discussion
Recently, pyridines and their fused ring systems have received considerable attention as potential chemotherapeutic agents. Among the wide range of pyridine derivatives, pyrano-pyridine family has been shown to possess high antitumor potency toward several types of cancer cell lines with noticeable activity against the leukemia cell line.
34
The cytotoxic activity of several thieno [2, 3-b] pyridine derivatives against MCF-7 and HepG-2 human cancer cell lines has been reported.
35
Anticancer activity of some 1,4-dihydropyridine containing nitroimidazole moieties on C4 position has also been evaluated against four different cancer cell lines.
36
In the present study, we report four compounds from this family that cause significant decrease in viability of the K562 cells in a dose- and time-dependent manner. These compounds were developed by substituting different group such as phenyl (P.P), thiophene (TPM.P), 4-chlorophenyl (4-CP.P), and 3-nitrophenyl (3-NP.P) in 4-position of phenyl ring (Figure 1). Substitution of the 4-chloro phenyl ring (4-CP.P) with phenyl, thiophene, and also 3-nitrophenyl groups increases its activity suggesting the presence of an electron withdrawing group (Cl) at the 4-position but not at the 3-position (NO2) of phenyl which is accompanied by a remarkable increase in cytotoxic effects of these compounds. However, the presence of another electron withdrawing group including NO2 (3-NP.P) at the 3-position reduces the activity of the compound (Table 1). This suggests that electron withdrawing groups at the 4-position of phenyl ring can be effective for increasing the cytotoxic activity of the compounds. Overall, the 3-nitro (3-NP.P) and other analogues (P.P, TPM.P) were significantly less potent than 4-CP.P (Tables 1 and 2). In addition, it was observed that the most active derivative (4-CP.P) could inhibit cancer cell proliferation via promotion of apoptosis by enhancing intracellular ROS generation, while simultaneously decreases the antioxidant factors levels. Recent studies implicate that the diverse chemotherapeutic agents can kill cancer cells by increasing oxidative stress. For instance, Trachootham et al. showed that β-phenylethyl isothiocyanate in transformed cells acted by depleting GSH pool and thus created stress conditions leading to cell death.
37
Moreover, adaphostin as a cytotoxic drug used in standard leukemia therapy is able to induce apoptosis in variety of BCR-ABL-expressing cells via increasing ROS following DNA damages.
38
With capability of reducing the viability in K562 cell line (Table 1), the results of apoptosis assessment in the present study (Figure 3) revealed the antitumor activities of mentioned compounds. According to the explained results and flow cytometry analysis, we propose that 4-CP.P induces apoptosis and tumor inhibition through ROS production and probably mitochondrial dysfunction.
39
The results of measuring the activity of enzymes confirm our statements (Figure 5). SOD is an essential enzyme for resolving superoxide radicals before they can participate in the formation of other ROS.
40
Due to death occurred under the condition of superoxide radicals high production and/or low SOD activity in malignant cells, these cells are dependent on SOD activity for survival. In this regard, it has been reported that indicated oestrogen derivatives can cause O2
·
Conclusion
In this study, we have reported four compounds of pyrano-pyridine derivatives with high antiproliferative and apoptotic inducing activity. Cellular oxidative damages and apoptosis induced by an active compound (4-CP.P) of these derivatives in K562 leukemia cell were reported. Our results showed that 4-CP.P induces overproduction of ROS, as well as lipid peroxidation, protein oxidation and total thiol depletion in K562 cells. The activity of SOD and CAT was also increased and decreased, respectively. These data suggest that 4-CP.P induces oxidative damage in K562 cells that ultimately triggers apoptosis.
Footnotes
Acknowledgments
The authors appreciate the support for this investigation by the research council of University of Tabriz, Tabriz, Iran. We would like to thank Dr. Sanam Arami for helping us in English editing.
Author contribution
S Asghari and M Rahnamay contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.