
Abstract
Previous histopathological studies have shown the hepatotoxicity of paclitaxel (TXL). However, there is little known about the molecular pathway(s) of TXL-induced hepatotoxicity. Therefore, this study aimed to uncover the role of two transcription factors in the TXL-induced hepatotoxicity. Moreover, the hepato-protective effect of royal jelly (RJ) on TXL-induced toxicity was investigated. Wistar rats were divided into control and test groups. The test groups along with TXL received various doses of RJ (0, 50, 100 and 150 mg/kg, body weight). Biochemical hepatic functional assays, histopathological studies and hepatic superoxide dismutase level were determined. Additionally, the expression of E2f1 and cellular-myelocytomatosis (c-Myc) at messenger RNA (mRNA) level in the liver was evaluated. The hepatic functional biomarkers showed a significant (p < 0.05) elevation in the TXL-received animals, while RJ administration for 28 days resulted in a remarkable reduction in TXL-elevated alkaline phosphatase, alanine transaminase and lactate dehydrogenase levels. The TXL-treated animals showed a significant (p < 0.05) up-regulation of E2f1 and down-regulation of c-Myc at mRNA level, respectively. RJ lowered the expression of E2f1 while enhanced the expression of c-Myc in a dose-dependent manner. Our data suggest the hepato-protective effects of RJ on TXL-induced toxicity, which may attribute to a clear crosstalk between E2f1 and c-Myc as two regulators of liver growth.
Introduction
During the last two decades, extensive research has been done on various signal transduction pathways, which are playing crucial role in cell fate decisions. Among the others, cellular-myelocytomatosis (c-Myc) and E2f1 are important regulators of cell signalling. Previous studies revealed that the c-Myc oncoprotein regulates up to 15% of all human genes that are essential in the regulation of cell behaviour, including cell growth and proliferation, differentiation and apoptosis. 1 The overexpression or deregulation of c-Myc has been reported in response to various factors such as hypoxia, DNA damage, glucose deprivation and anticancer agents. 2 c-Myc as a transcription factor when heterodimerized with the Max polypeptide can bind to a DNA sequence and activate the expression of genes, which are important for cell cycle progression and metabolism. 3
E2f1 is also a transcription factor required for cell proliferation and growth by binding to the promoter region of several genes, including those that are involved in cell cycle regulatory activities and DNA replication. 4 In addition to cell proliferation, E2f1 can induce apoptosis when it is overexpressed or deregulated through Rb inactivation. 5 Both E2f1 and cMyc do have some regulatory properties in common including deregulated expression of both transcription factors can stimulate quiescent cells to enter the S phase of the cell cycle and either factors act dually in cell proliferation and apoptosis. On the other hand, in spite of some regulatory similarities between these two transcription factors, there are different reports suggesting that c-Myc may be downstream of E2f1, upstream of E2f1, or even functions in a parallel pathway with E2f1 to regulate cell proliferation. 6 However due to the fact that E2f1 DNA-binding sites are found in the c-Myc gene promoter, thus at least under some conditions, these sites are responsive to E2f1-dependent transcription. 7 There are other reports about using developed transgenic models in which overexpression of E2f1 and/or c-Myc was targeted at the liver. The mentioned investigations showed that the combined expression of these two transcription factors remarkably accelerated hepatic cell carcinoma growth compared to either E2f1 or c-Myc mono-transgenic mice. 8
Paclitaxel (Taxol (TXL)) as an effective chemotherapeutic agent and mitotic inhibitor is used against wide range of solid tumours such as ovarian, breast, lung and prostate cancers. It promotes the polymerization of tubulin and interposes with microtubule depolymerization, which is essential in cell division. Likewise other chemotherapy agents TXL also do have side effects such as neuropathy, endothelial dysfunction, cardiotoxicity, hypersensitive reactions and gastrointestinal dysfunction, which diminish its high effectiveness. 9 –11 At the same time, TXL-induced hepatotoxicity and the protective effect of β-1, 3-D-glucan, against liver damage induced by TXL has been investigated. 12 Moreover, implication of HIF-1 and AP-1 transcription factors in the protective role of hypoxia against the TXL-induced apoptosis in MDA-MB-231 breast cancer cells has been reported. 13
Royal jelly (RJ), as hypopharyngeal glands secretion of nurse bees, is an exclusive food for the queen honey bee (apismillifera) larva. RJ consists of water (50–60%), proteins (18%), carbohydrates (15%), lipids (3–6%), mineral salts (1.5%) and vitamins. Additionally, RJ contains many bioactive substances such as 10-hydroxyl-2-decenoic acid with immunomodulating properties, antibacterial protein, fatty acids and peptides. RJ has plenty of pharmacological properties including antioxidant capacity, antimicrobial activities, insulin-like effect, antitumour activity, vasodilatotary activity, antihypercholesterolemic, antihypertensive, antiallergic, antifatigue, wound-healing properties and protective effect against hematopoietic dysfunction. 14,15
Since the hepatotoxicity of TXL has been previously reported from histopathological point of view, we in this study aimed to show the possible protective effect of RJ on TXL-induced histopathological and biochemical alterations in the liver. Additionally, we also aimed to dissect out any changes in the expression of two important transcription factors E2f1 and c-Myc at the messenger RNA (mRNA) level in the liver of TXL-exposed non-treated and RJ-treated animals.
Materials and methods
Chemicals
5.5′-Dithiobis-2-nitrobenzoic acid and guanidine hydrochloride were purchased from Sigma-Aldrich (Germany). Thiobarbituric acid, phosphoric acid (85%), trichloracetic acid, dimethyl sulfoxide and ethanol were obtained from Merck (Germany). N-butanol was obtained from Carl Roth, GmbH Co. (Germany). TRIzol reagent was purchased from Applied Biosystems by life technologies (Nieuwerkerk, the Netherlands). Commercially available standard kits were used for the determination of alkaline phosphatase (ALP, 744; Man Inc., Tehran, Iran), alanine transaminase (ALT, 1-400-019; ParzAzmun, Tehran, Iran), lactate dehydrogenase (LDH, 10-533-1; Ziest Chem Diagnostic, Tehran, Iran) and superoxide dismutase (SOD, Ransol; RANDOX laboratories Ltd, Crumlin Co. Antrim, UK). All other chemicals were commercial products of analytical grade.
RJ was collected from beehive nos. 8 and 14 (Sardrood, Hamadan province, Iran), during 2013 and kept at −20°C until use. RJ was dissolved in distilled water and given orally.
Animals and experimental design
Forty-eight adult male Wistar rats (200–220 g) were obtained from the animal resource of the Faculty of Veterinary Medicine, Urmia University. The rats were examined and their healthiness was confirmed by a veterinarian who works in pet animal field. The animals were acclimatized for 1 week and had free access to food and water. The experimental protocols were approved by the ethical committee of Urmia University in accordance with principles of laboratory animal care. Animals were assigned into control (C) and test (T) groups (n = 8). Animals in the T group were subdivided to the following groups:
TXL group received TXL (7.5 mg/kg bw, intraperitoneally, each 7 days); T1 group along with TXL received RJ (50 mg/kg bw, at 11:00 am, daily); T2 group along with TXL received RJ (100 mg/kg bw, at 11:00 am, daily); T3 group along with TXL received RJ (150 mg/kg bw, at 11:00 am, daily); T4 group received only RJ (100 mg/kg bw, at 11:00 am, daily).
The control group received only saline (0.9%, 5 ml/kg) during the experiment. All T groups received RJ and TXL for 4 weeks. The RJ dose levels were selected based on previous reports and our primary pilot experiments. 16 To evaluate any treatment-related changes in the body weight gain, all animals were weighed individually before and after the experimental procedures.
Serum preparation and tissue samples collection
On day 29, blood samples were obtained by cardiac puncture under light anaesthesia, which was provided using diethyl ether. After keeping for 10 min at room temperature, the samples were centrifuged at 3000g for 10 min to obtain the serum. The serum samples were then stored at −20°C until further analyses.
The anaesthetized animals were ultimately euthanized using CO2 gas. The liver specimen was immediately removed and rinsed with chilled saline. A small piece of the liver samples preferably the same location from each individual rat was snap frozen in liquid nitrogen and kept at −70°C until further biochemical and molecular analyses and the rest of each single sample was fixed in formalin solution (10%) for histopathological examinations.
Determination of the serum level of hepatic functional enzymes
The serum levels of ALT and ALP were measured using commercially available standard kits according to the manufacturer’s instructions.
Lactate dehydrogenase and superoxide dehydrogenase assessment in the liver
The hepatic concentrations of LDH and SOD were determined using LDH (12-506; Ziest Chem Diagnostics) and SOD (Ransol; RANDOX laboratories Ltd, Crumlin Co.) kits and according to the manufacturer’s instructions.
Estimation of lipid peroxidation
To determine the lipid peroxidation rate, malondialdehyde (MDA) content of collected liver samples was measured using the thiobarbituric acid (TBA) reaction as described previously. 17 In short, 0.2–0.3 g of the liver samples was homogenized in ice-cold potassium chloride (KCl) (150 mM), and then the mixture was centrifuged at 3000g for 10 min. Thereafter, 0.5 ml of the supernatant was mixed with 3 ml phosphoric acid (1% v/v) and then following vortex mixing, 2 ml of 6.7 g L−1 TBA was added to the samples. The samples were heated at 100°C for 45 min and chilled on ice. Finally, 3 ml N-butanol was added and the samples were further centrifuged at 3000g for 10 min again. The absorbance of supernatant was measured spectrophotometrically at 532 nm and the MDA concentration calculated according to the simultaneously prepared calibration curves using MDA standards. The amount of MDA was expressed as nanomoles per milligram protein of the samples. The protein content of the samples was measured according to the Lowry method. 18
Protein carbonylation assay (protein oxidation)
To determine the carbonyl content of the liver homogenates, the reaction between 2, 4-dinitrophenylhydrazine (DNPH) and protein carbonyls was measured. 19 Briefly, 0.2–0.3 g of the samples were homogenized in ice-cold phosphate buffer (50 mM, pH 6.7 and containing 1 mM EDTA), and then the mixture was centrifuged at 10,000 × g for 10 min at 4°C. For each individual sample (0.2 ml supernatant), the T and C samples were prepared and then 0.8 ml of DNPH and 2 M HCl solution were added to the T and C samples, respectively. The samples were kept in dark at room temperature for 1 h, with vortex mixing every 15 min. Thereafter, 0.5 ml trichloracetic acid (30%) were added in each sample and vortex mixed for 30 s. All samples were centrifuged at 10,000g for 3 min, the supernatant was discarded and the precipitate resuspended for 15 min in 1 ml of (1:1) ethanol/ethyl acetate solution. After centrifugation at 10,000g for 3 min and discarding the supernatant, the above step was repeated. Following the last wash, the precipitates were dissolved with 0.6 ml guanidine hydrochloride solution (6 M) at 37°C for 15 min. After dissolving the precipitate, the samples were centrifuged at 10,000g for 3 min to get rid of any leftover debris. For each sample of T and C, the optical density (OD) was measured against 6 M guanidine hydrochloride solution at a wavelength of 370 nm.
The carbonyl content was determined as follows:

where CA is the corrected absorbance and computed as the average OD for each control sample, which was subtracted from average OD of T sample at 370 nm. The extinction coefficient for DNPH at 370 nm is 22,000 M−1 cm−1. In order to determine the carbonyl content per milligram of protein, the protein levels were measured at 280 nm in each sample.
Measurement of total thiol molecules
Total sulphydryl level in the liver samples was measured as described previously. 20 Briefly, 0.2–0.3 g of the liver samples was homogenized in ice-cold KCl (150 mM), and the mixture was centrifuged at 3000g for 10 min. To 0.2 ml of the supernatant of the tissue homogenate, 0.6 ml Tris-ethylendiamintetra-acetic acid buffer (Tris base 0.25 M EDTA 20 mM, pH 8.2) and thereafter 40 μl 5.5′-dithiobis-2-nitrobenzoic acid (10 mM in pure methanol) were added. The final volume of this mixture was made up to 4.0 ml by the addition of pure methanol. After 15 min incubation at room temperature, the samples were centrifuged at 3000g for 10 min and ultimately the absorbance of the supernatant was measured at 412 nm. The total thiol molecule (TTM) capacity was expressed as nanomoles per milligram of protein in samples. The protein content of the samples was measured according to Lowry et al.
Nitric oxide measurement
The total nitrate/nitrite content of the liver samples was measured according to the Griess reaction. 21 In Griess reaction, nitric oxide (NO) is rapidly converted into more stable nitrite, and in acidic environment the nitrite is converted into nitrous acid (HNO2). In reaction with sulphanilamide, HNO2 forms a diazonium salt, which reacts with N-(1-naphthyl) ethylenediamine.2HCL to form an azo dye that can be detected by at 540 nm wavelength. The NO content in the liver is expressed as nanomoles per milligram of protein.
Quantitative DNA fragmentation assay
The DNA fragmentation quantified spectrophotometrically according to the previously described method. 22 In short, a small piece of the frozen liver sample was homogenized in chilled lysis buffer (Tris-HCl/EDTA/Triton X-100; pH 8.0). Homogenates were centrifuged at 27,000g for 20 min to separate intact chromatin in the pellet from and fragmented DNA in the supernatant. Pellets and supernatants were thereafter treated with perchloric acid (0.5 N), boiled at 90°C for 15 min and centrifuged at 1500g for 10 min to precipitate protein and other debris. Resulting supernatants were then treated with Burton’s diphenylamine reagent for 16–20 h at room temperature in the dark. Absorbance was measured at 600 nm wavelength. DNA fragmentation in samples after adjusting based upon their individual weight was expressed as percentage of total DNA appearing in the supernatant fraction. Treatment effects are reported as percentage of control fragmentation.
RNA isolation and reverse transcription polymerase chain reaction
To evaluate the effect of RJ on the expression of E2f1and c-Myc at mRNA level, total RNA was isolated from the liver samples using the standard TRIZOL method. 23 To avoid genomic DNA contamination, extra care was taken when the colourless aqueous phase collected after chloroform extraction. The RNA amount was determined spectrophotometrically (260 nm and A260/280 = 1.8–2.0), and the samples were stored at −70°C. For reverse transcription polymerase chain reaction (RT-PCR), complementary DNA (cDNA) was synthesized in a 200-μl reaction mixture containing 1 µg RNA, oligo(dT) primer (1 µl), 5× reaction buffer (4 µl), RNAse inhibitor (1 µl), 10 mM dNTP mix (2 µl) and M-MuLV reverse transcriptase (1 µl) according to the manufacturer’s protocol (Fermentas, GmbH, Germany). The cycling protocol for 20 μl reaction mix was 5 min at 65°C, followed by 60 min at 42°C and 5 min at 70°C to terminate the reaction.
Second strand cDNA synthesis
The RT-PCR reaction was carried out in a total volume of 25 µl containing PCR master mix (12.5 μl), forward- and reverse-specific primers (each 0.75 μl) and cDNA as a template (1 µl) and nuclease free water (10 µl). PCR conditions were run as follows: general denaturation at 95°C for 3 min, 1 cycle, followed by 35 cycles of 95°C for 20 s; annealing temperature (63°C for Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 58°C for E2f1and 59°C for c-Myc) for 30 s; elongation: 72°C for 1 min and 72°C for 5 min.
The products of RT-PCR were separated on 1.5% agarose gel containing ethidium bromide and visualized using Gel Doc 2000 system (Bio-Rad). The specific primers for Ratus E2f1, c-Myc and GAPDH were designed and manufactured by CinnaGen (CinnaGen Co. Tehran, Iran). 24 –26 Primers pairs for RT-PCR are listed in Table 1. Densitometric analyses of PCR products were performed using Molecular Analyst software (version 1.5) from BioRad (Hercules, California, USA).
neuclotid sequences, products size and anneling tempratures for primers used in RT-PCR.

RT-PCR: reverse transcription polymerase chain reaction.
Histopathological examinations
Previously fixed liver samples were subjected to histopathological examinations. The samples were embedded in paraffin and sections (5–6 µm) were stained with haematoxylin and eosin and analysed under light microscope by multiple magnifications. To evaluate the level of damages following exposure to TXL and/or TXL plus RJ treatments, indices such as cell swelling, congestion and necrosis in the liver were scored, numerically. The evaluation criteria were as follows: zero for no detectable lesion, 1 for mild changes, 2 for moderate changes and 3 for severe damages. For each animal in the T and C groups, at least three slides were prepared and scored.
Statistical analyses
For all numerical results, mean and standard deviation of the measured parameters were calculated. The results of three independent experiments for each assessment were analysed using Graph Pad Prism software (version 2.01; Graph Pad software Inc. San Diego, California). The comparisons between groups were made by analysis of variance followed by Bonferroni post hoc test. For comparing the graded degree of pathological findings between groups, the Kruskal–Wallis test was used. A p- value <0.05 was considered significant.
Results
RJ normalized the TXL-elevated serum level of hepatic functional enzymes
Serum level of hepatic functional enzymes and LDH as a marker for tissue damage was assessed and all three biomarkers showed a significant (p < 0.05) elevation in the TXL-received animals. Administration of RJ for 28 days resulted in a remarkable and dose-dependent reduction of TXL-elevated ALP, ALT and LDH levels. Those animals that received only RJ did not show any significant changes when compared with the control group (Table 2).
Effects of RJ on TXL-elevated serum level of ALP, ALT and LDH (U/L).a
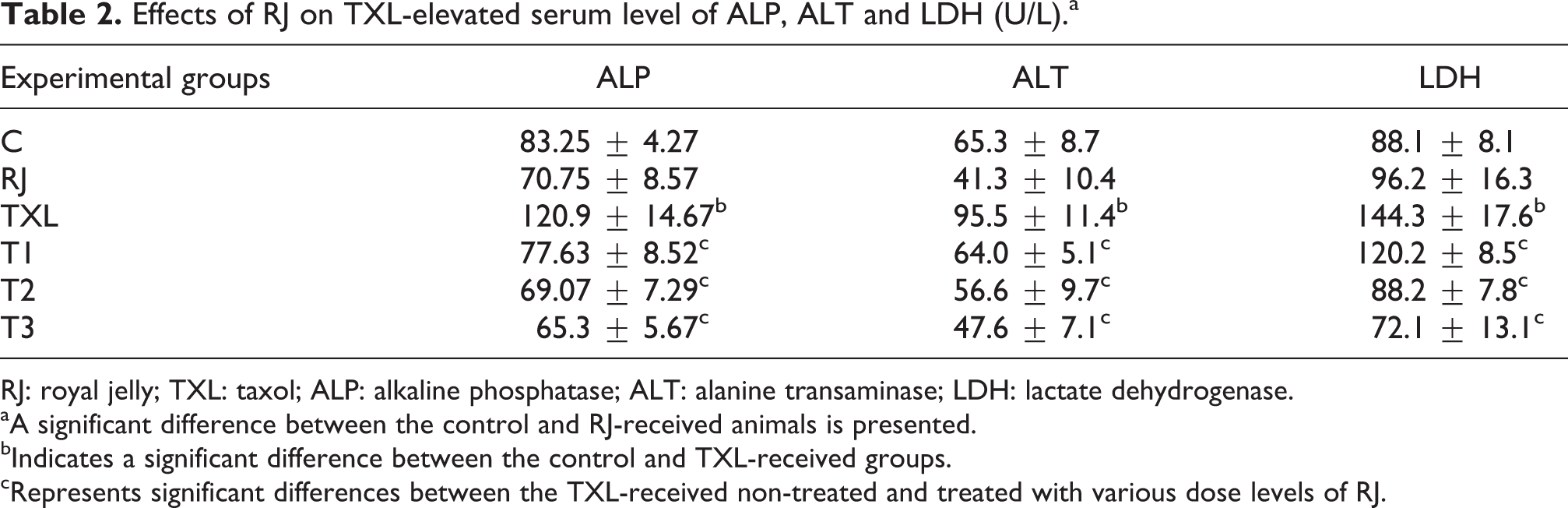
RJ: royal jelly; TXL: taxol; ALP: alkaline phosphatase; ALT: alanine transaminase; LDH: lactate dehydrogenase.
aA significant difference between the control and RJ-received animals is presented.
bIndicates a significant difference between the control and TXL-received groups.
cRepresents significant differences between the TXL-received non-treated and treated with various dose levels of RJ.
RJ protected from TXL-induced lipid and protein oxidation
The concentration of MDA as end product of lipid peroxidation and the level of carbonylated protein as biomarker of protein oxidation in the liver tissue were measured. Either biomarker indicated that the TXL-received animals show marked increase in lipid and protein oxidation (Figure 1(a) and (b)). RJ administration resulted in a significant (p < 0.05) and dose-dependent protection from TXL-induced protein carbonylation, while the strongest anti-lipid peroxidation effect of RJ was obtained at medium given dose level (Figure 1(a)). Although RJ alone could elevate the lipid and protein oxidation, the increased levels were not statistically significant (p > 0.05).

Effect of RJ on TXL induced: (a) MDA level, (b) cabonylated protein level in the liver; data are given as mean ± SD (n = 8). Stars indicate a significant difference between the control and TXL-received groups and # represents significant differences between the TXL-received non-treated and treated with various dose levels of RJ. A significant difference between the control and RJ-received animals is presented by $. RJ: royal jelly; TXL: taxol; SD: standard deviation.
RJ balanced the TXL-imbalanced oxidative and nitrosative stress
To evaluate the hepatic level of enzymatic antioxidant capacity, SOD activity was measured. Results indicated that the TXL administration lowered the SOD activity significantly (p < 0.05), while those TXL-treated animals that received various dose levels of RJ showed marked protective effect of RJ on TXL-induced SOD activity reduction. The highest SOD activity was found in the T2 group, which received medium dose of RJ. Administration of RJ alone increased the hepatic SOD activity significantly (Figure 2(a)). NO content of the liver was assessed as a biomarker of nitrosative stress and the results showed that the TXL-received untreated group demonstrated the highest NO concentration in the liver. The RJ administration alone did not result in a statistical change in the hepatic concentration of NO, while in the TXL-received animals lowered the NO content and the lowest concentration of NO was found in the rats which were treated with medium dose of RJ (Figure 2(b)).

Effect of RJ on TXL induced: (a) superoxide dismutase activity and (b) NO content in the liver; data are given as mean ± SD (n = 8). Stars indicate a significant difference between the control and TXL-received groups and # represents significant differences between the TXL-received non-treated and treated with various dose levels of RJ. A significant difference between the control and RJ-received animals is presented by $. RJ: royal jelly; TXL: taxol; SD: standard deviation; NO: nitric oxide.
TXL-induced DNA fragmentation was reduced by RJ administration
Influence of RJ on TXL-induced genomic DNA fragmentation in the liver was evaluated by quantitative method. DNA fragmentation was quantitated at 24 h by differential centrifugation, sedimentation and reaction with Burton’s reagent. Very slight but significant fragmentation (11%) was found in the TXL-received group, which remarkably was reduced in the RJ-treated groups (Figure 3).

Effect of RJ on TXL-induced DNA fragmentation in the liver; data are given as percentage of control. *The value is significantly different from vehicle-treated control and TXL-received animals. # is significantly different from untreated TXL-received animals. RJ: royal jelly; TXL: taxol; SD: standard deviation.
RJ improved the TXL-induced histopathological injuries
Histopathological examinations showed a normal and intact architecture of the liver in the control and RJ-received groups (Figure 4(a) and (b)). Hepatocytes swelling, necrosis and destruction of nuclei, mononuclear cell infiltration and congestion were the pathological injuries in the liver of TXL-received animals (Figure 4(c)). Although the low-dose of RJ was not able to reduce the TXL-induced hepatic injuries, medium and high given dose levels relatively and remarkably improved the damages, respectively (Figure 4(d) and (e)). The numerically scored parameters have been depicted in Table 3.
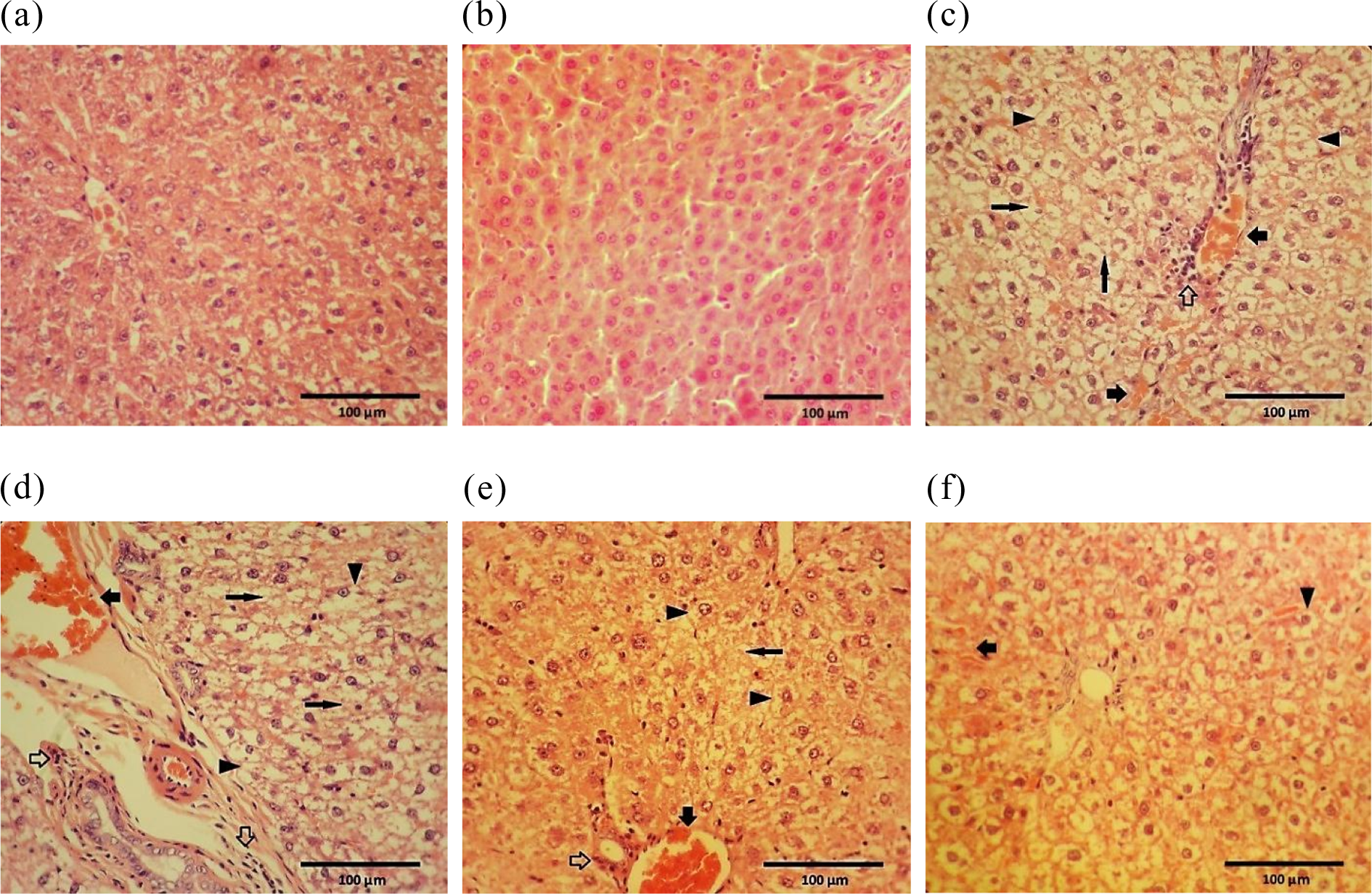
Effect of RJ on TXL-induced Histopathological injuries in the liver: Control (a); RJ-received animals (b), TXL-treated group (c) to (f) are representing the liver sections from animals that received low, medium and high dose levels of RJ along with TXL, respectively. Severe cell swelling (arrow head), cell necrosis and destruction of nuclei (arrow), limited congestion (bold arrow) and mononuclear cell infiltration (hollow arrow) have been labelled. Haematoxylin and eosin staining; original magnification, ×100 and scale bars = 100 µm. RJ: royal jelly; TXL: taxol; SD: standard deviation.
Effect of RJ on histological changes in the liver of TXL-exposed rats; mean values ± SD are given.

RJ: royal jelly; TXL: taxol; C: control; T: test.
aA significant difference between the control and RJ-received animals is presented.
bA significant difference between the control and TXL-received groups.
cSignificant differences between the TXL-received non-treated and treated with various dose levels of RJ.
RJ regulated the TXL-altered expression of E2f1 and c-Myc in the liver
The mRNA level of E2f1 and c-Myc was determined using a semi-quantitative PCR technique and the results were normalized against the mRNA level of GAPDH as a house-keeping gene (Figure 5(c)). RJ alone did not change the E2f1 expression while remarkably down-regulated the expression of c-Myc in the liver. The TXL-treated animals showed a significant (p < 0.05) up-regulation of E2f1 and down-regulation of c-Myc at mRNA level, respectively. RJ administration lowered the expression of E2f1 and enhanced the expression of c-Myc in a dose-dependent manner (Figure 5(a) and (b)). The density of both genes in the liver was measured by densitometry and normalized to GAPDH mRNA expression level and was expressed as integrated density values of E2f1 and c-Myc mRNA levels (Figure 5(d) and (e)).

Effect of TXL and RJ on: (a) E2f1 (198 bp), (b) c-Myc (479 bp) and (c) GAPDH (167 bp) mRNA levels in the liver, the expression of E2f1, c-Myc and GAPDH at mRNA level were evaluated by semi-quantitative RT-PCR. The densitometric estimations were conducted and panels (d) and (e) are representing the density of E2f1 and c-Myc mRNA in the liver that were normalized to GAPDH mRNA expression level. Results were expressed as IDV of E2f1 and c-Myc mRNA levels. RJ: royal jelly; TXL: taxol; SD: standard deviation; RT-PCR: reverse transcription polymerase chain reaction; IDV: integrated density value.
Discussion
This study showed that RJ could protect from the TXL-induced hepatic damages, in which the hepato-protective effects of RJ were characterized by reversing the elevated hepatic functional enzyme level, reducing the TXL-induced lipid and protein oxidation, improving the TXL-induced pathological injuries and lowering the TXL-induced DNA fragmentation in the liver. Moreover, the RJ treatment resulted in different effects on the expression of E2f1 and c-Myc; as the expression of E2f1 dose dependently was down-regulated, while the expression of c-Myc was up-regulated.
Although bone marrow suppression has been reported as the major toxicity of TXL, there are however emerging data indicating its toxic effects in other organs including neuronal system and the liver. 27 Therefore, in this study, we primarily aimed to highlight the TXL-induced hepatotoxicity by hepatic structural and functional assays including the measurement of serum level of ALT and ALP and hepatic content of LDH. Indeed, a significant increase in serum concentration of these biomarkers indicated the TXL-induced hepatoxicity, representing the TXL-induced hepatocytes and also bile duct dysfunction, hepatocyte membrane disintegration, respectively.
The main mechanism of anticancer action of TXL has been related to its ability to cause abnormal stabilization of the dynamic microtubule polymerization and consequently to the failure of mitosis. Nevertheless, other mechanisms such as alteration of intracellular signalling and organelle transport, associated with microtubules, also play crucial role in its anticancer property. 28 Previous studies also showed that TXL was able to produce reactive oxygen species in human breast cancer (MCF-7) cells, which among the others hydrogen peroxide was found the main cause of cancer cell death. 29,30 Our findings concerning the TXL-induced oxidative stress in the liver confirmed the previous evidence and extended the fact that TXL is able to induce oxidative stress in normal hepatocytes too. The elevated hepatic level of lipid and protein oxidation end products (MDA and carbonylated protein, respectively) along with mild decline in hepatic SOD level represented a marked imbalance in antioxidant system of the liver. We also found that the TXL-induced stress is not limited to oxidative stress as the hepatic concentration of NO was found remarkably elevated in the TXL-received animals.
Our findings are indicating that the mechanism of cell death in the liver following the TXL-treatment is necrosis rather than apoptosis as morphologically cytoplasmic swelling and destructed nuclei in hepatocyte in histopathological examinations and very slight DNA fragmentation represent that most of the cell injuries might be categorized as necrotic damages. There are supporting data indicating that TXL by two different mechanisms causes apoptosis and necrosis. At lower concentrations (0.005–0.05 μM), TXL causes stabilization of the spindle during mitosis that led to the inhibition of proliferation and the induction of apoptosis, while at higher concentrations (5–50 μM), TXL increases the polymerization of microtubules and stimulates the formation of microtubule bundles that blocks entry into S phase and ultimately the induction of necrosis. 31 Another study using the Annexin V and PI methods in human leukemic U937 cells showed that after the TXL treatment, the cells in G1 and S stages died only because of apoptosis, whereas G2/M-stage cells died because of both apoptosis and necrosis. The authors also demonstrated that like our findings, the TXL-treated cells released significant amount of LDH, indicating cell membrane disruption, suggesting the capability of TXL to induce both apoptosis mainly in S stage and necrosis in the G2/M stages. 32
Until now, nine E2F family members have been identified (E2f1-8 with E2f3 having two variants: E2f3a and E2f3b) and certain E2fs are associated with either transactivation or repression of target genes. E2f1, E2f2 and E2f3a are commonly referred as the ‘activating’ E2fs due to their capability to activate the transcription of genes. Among the nine members of the E2f family, E2f1 is unique in its ability to induce apoptosis. 33 At the same time, the c-Myc gene encodes the transcription factor c-Myc, which heterodimerizes with Max, to regulate gene expression. C-Myc as an oncogenic transcription factor that integrates the cell cycle machinery with cell adhesion, cellular metabolism, and the apoptotic pathways. 34 There is a close crosstalk between c-Myc and E2f1 signalling pathway, which is essential to control cell fate decisions. 35 We found that the TXL treatment resulted in a remarkable up-regulation of E2f1 and by contrast down-regulation of c-Myc in the liver. Previous studies showed that c-Myc and E2f1 can activate each other’s transcription, which results in a positive feedback circuit, while our results in the liver of TXL-treated animals are not supported with this positive relationship. 36 The reason for this discrepancy might be related to the unique cell proliferation processes in the liver, which does have distinct cell multiplication, cell polyploidization and cytoplasmic hypertrophy. This is in contrast with preneoplastic growth that mainly is dominated by a mono-nucleated diploid cell population. 37
The second part of this study devoted to uncover any beneficial effects of RJ on the TXL-induced hepatic injuries. Our findings indicated that RJ in a dose-dependent fashion could protect from the TXL-induced hepatotoxicity. The RJ co-administration with TXL for 28 days not only improved the hepatic functional-related markers in serum including ALP, ALT and LDH but also improved the oxidative and nitrosative stress biomarkers. Previous studies already demonstrated the hepato-protective effect of RJ on paracetamol-induced hepatotoxicity in rats. 38
RJ along with anti-lipid peroxidation and anti-protein oxidation capacities enhanced the SOD activity, suggesting its dual antioxidant properties. The antioxidant effect of RJ has been shown on fumonisin- and cisplatin-induced oxidative stress. 39,40 Recently, anti-lipid peroxidation effect of RJ has been reported in azathioprine-induced lipid peroxidation in the liver. 41 We found no previous report indicating the hepato-protective effect of RJ on the TXL-induced hepatotoxicity. Although there is no precise known mechanism of hepato-protective for RJ, but as early studies have shown it does contain biologically active substances such as aspartic acid, cystine, cysteine, tyrosine and glycine, which directly or indirectly playing a hepato-protective role. 38 For example, cystine and cysteine are essential amino acids that participate in the synthesis of glutathione peroxidase, a known antioxidant enzyme. Additionally, it has been frequently reported that glycine is one of the best antioxidants to encounter the heavy metal–induced oxidative stress. 42 As mentioned above, the most published research during the last decade indicate that RJ exert its beneficial effects through its antioxidant properties. 39 Moreover, RJ does contain a glycoprotein with 57-kDa weight, which stimulates hepatocyte development and liver regeneration. 43 As RJ administration markedly declined the TXL-elevated NO level, thus it may be suggested that RJ in addition of antioxidant capacity via inhibition of nitrosative stress also play hepato-protective role. Anti-nitrosative effect of RJ has been reported in the reduction of secondary neuronal damage in rabbits. 44
Our findings show an opposing behaviour in the expression of two transcriptional factors, where following the TXL treatment, expression of E2f1 was up-regulated, while the expression of c-Myc was down-regulated. At the same time, those groups of animals, which were co-treated with TXL and RJ also showed an opposing feature in the expression of either gene. Although E2f1 and c-Myc do have some common regulatory properties but no functional, relationship between these two has been reported.
There are numerous reports indicating that E2f1 provokes apoptosis both in in vitro condition such as TNF-α or doxorubicin-induced apoptosis in osteosarcoma cell line and in in vivo models such as abnormal accumulation of T lymphocytes in thymus of E2f1-null mice. 5 On the other hand, it has been reported that the expression of c-Myc is induced by growth factors and in turn results in entering the quiescent cells into S phase of cell proliferation cycle. 45 Although our results did not dig the detail mechanisms which how RJ at the same time can down and up-regulate the E2f1 and c-Myc genes at mRNA level but apparently RJ attenuated the TXL-induced E2f1 and consequently cell death and elevated the cell proliferation by up-regulation of c-Myc in the liver. There are several supporting reports indicating a crosstalk mechanism between these two transcription factors. For example, it has been shown that the c-Myc-mediated proliferation and lymphomagenesis were compromised by E2f1 loss. 46 In another study, an inhibition property of E2f1 and acceleration capacity of c-Myc on hepatic ploidy and tumorigenesis in transgenic mouse models have been documented. 47
Conclusion
We showed that TXL at tested dose level (7.5 mg/kg, weekly) can result in hepatic injuries, which were characterized by marked alterations in hepatic functional enzymes concentration, oxidative stress markers elevation, histopathological changes and ultimately by up- and down-regulation of E2f1 and c-Myc genes at mRNA level. All aforementioned factors were improved in those animals which co-treated with TXL and RJ. We found a clear crosstalk between E2f1 and c-Myc as two critical regulators of liver growth and believe that RJ exerts its potential hepato-protective effects via its antioxidant capacity and also acting as a growth factor, which is enable to mediate the expression of E2f1 and c-Myc in the liver.
Footnotes
Authors’ Contribution
HM designed the study, conducted the experiments, analysed the data, discussed and wrote the manuscript. MF and FDK conducted the experiments. SKSR and ARG discussed and analysed data. All authors approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by Urmia University (project no: 007/D/92).