
Abstract
Predictive toxicology plays a critical role in reducing the failure rate of new drugs in pharmaceutical research and development. Despite recent gains in our understanding of drug-induced toxicity, however, it is urgent that the utility and limitations of our current predictive tools be determined in order to identify gaps in our understanding of mechanistic and chemical toxicology. Using recently published computational regression analyses of in vitro and in vivo toxicology data, it will be demonstrated that significant gaps remain in early safety screening paradigms. More strategic analyses of these data sets will allow for a better understanding of their domain of applicability and help identify those compounds that cause significant in vivo toxicity but which are currently mis-predicted by in silico and in vitro models. These ‘outliers’ and falsely predicted compounds are metaphorical lighthouses that shine light on existing toxicological knowledge gaps, and it is essential that these compounds are investigated if attrition is to be reduced significantly in the future. As such, the modern computational toxicologist is more productively engaged in understanding these gaps and driving investigative toxicology towards addressing them.
Keywords
Introduction
The ability to predict the toxicological side effects of new chemical entities is critical to improving the efficiency of costly drug discovery.
1
Many predictive tools used in the current safety paradigms were designed to recognize risky compounds early in the drug discovery process, enabling a ‘fail early’ strategy. Examples include fundamental toxicity assays based on cellular adenosine triphosphate depletion (‘cytotoxicity’), mitochondrial dysfunction and glutathione depletion.
2
These tools have largely been developed from an understanding of the toxicological mechanisms of drugs or chemicals that have been withdrawn from the market for safety reasons. For example, nefazodone (

Metabolic oxidation of nefazodone
The metabolic liability of the chloroaniline moiety, coupled with the prescribed, large daily dose of >200 mg/day, 6 potentially exposes a metabolically active system to undesired levels of covalent adducts and marked reductions in the cellular antioxidant, glutathione, a mechanism that is proposed to lead to serious organ toxicities. 7
The ability of in vitro toxicity assays to assess or characterize the in vivo safety liabilities of toxic drugs such as nefazodone is sufficient justification for their inclusion in early safety screening cascades. Yet, despite the recent advances in predictive toxicology, the number of compounds failing in the clinic for unanticipated off-target toxicity remains unacceptably high. A recent example is ximelgatran, an antithrombotic and anticoagulant prodrug that was withdrawn in 2006 owing to liver toxicity. Subsequent reports suggest that ximelgatran induces a delayed hypersensitivity reaction, indicating an immunological role, but the underlying mechanism remains unclear and there were no assays at that time developed to provide early signals of this liability in the preclinical or clinical screening. 8 Withdrawn drugs such as ximelagatran are important to investigate because they expose toxicological knowledge gaps in the current safety screening paradigms.
If we are to continue to improve the prediction of toxicity, there is an urgent need to identify gaps in our current safety screening cascades. This can be done through understanding, at a structural and mechanistic level, the chemical and pharmacological space used to validate in vitro assays and train in silico models. Developing an analytical process to provide this understanding will help define the applicability domain of each assay or model, will expose mis-predicted compounds and thus help identify the toxicological knowledge gaps. These gaps can then drive investigative toxicology to focus on high-value chemistry spaces or mechanistic liabilities that are not adequately predicted by current safety paradigms. These efforts are critical to the development of effective safety screening cascades and the in vitro assays and in silico strategies that are used within them.
In silico modelling of in vivo data
The development of in silico models for the prediction of well-defined in vitro toxicological end points, such as Ames mutagenicity and uncoupling of oxidative phosphorylation, is largely successful owing to the relatively small number of simple mechanisms that underpin activity in these types of end points, that is, respectively, electrophilic reactivity towards DNA 9 and the presence of a lipophilic protonophore. 10 In contrast, for in vivo toxicological end points, the development of useful in silico models is far more challenging because in vivo toxicity is mechanistically complex. For example, nefazodone has liabilities in several in vitro assays, as previously discussed, and it is a challenge to determine which mechanism or mechanisms contribute to its hepatotoxic profile.
There are two factors that are driving the observation of in vivo toxicological events caused by drugs and drug candidate molecules: (1) drug exposure at the site or sites of action and (2) the ‘toxicological potential’ of the drug, that is, the ability or available weaponry of any molecule to cause damage in an in vivo system robustly evolved to withstand toxicological assault. In the absence of adequate exposure or sufficient toxicological potency, the likelihood of a toxic response is low. The dependency on drug exposure along with the multitude of mechanisms that may contribute to in vivo toxicity makes building generic computational toxicity models extremely challenging. As a consequence, machine learning algorithms generally capture physicochemical descriptors, applicable across chemical space, which predict exposure rather than true toxic potential.
For instance, in 2008, Hughes and coworkers 11 undertook a large-scale regression analysis employing physicochemical and structural descriptors to differentiate preclinical drug candidates that were annotated with respect to their in vivo toxicity. Specifically, compounds were labelled toxic if adverse in vivo observations were found at total compound plasma exposures of less than 10 µM and were labelled non-toxic if no adverse observations were present at this threshold. The results from their analysis yielded two physicochemical properties and associated thresholds beyond which the likelihood of seeing in vivo toxicities in preclinical candidates was significantly increased. To summarize their conclusions, compounds with a calculated lipophilicity (ClogP) >3 and a total molecular polar surface area (TPSA) <75 were almost six times more likely to correlate with observed in vivo toxicity when compared to compounds not crossing these thresholds (Table 1).
Hughes’ data set toxicity odds ratios observed for ClogP vs. TPSA (N compounds).

TPSA: total molecular polar surface area.
In contrast, Muthas and coworkers 12 published an analysis of 150 candidates from preclinical and phase I studies, classified according to success in their development milestones and found that the reverse was true, that is, compounds with a ClogP < 3 and a TPSA > 75 were more likely to cause observable toxic events in vivo. Although these studies were derived on compounds from different companies (i.e. Pfizer vs. AstraZeneca), and at different stages of the drug discovery process (i.e. preclinical vs. clinical), they expose disconnects between the two studies, and their utility is challenged in the absence of an analysis that defines their applicability.
Offering an objective analysis, Tarcsay and Keserű 13 reviewed the contribution of various physicochemical properties towards describing compound promiscuity, which is often associated with increased toxic potential and drug side effects, in data sets derived from several pharmaceutical companies, namely AstraZeneca, GlaxoSmithKline, Pfizer, Merck and Roche. Although they found a positive relationship between log P and promiscuity in all company data sets, compounds from AstraZeneca and GlaxoSmithKline yielded a positive correlation with molecular weight (MW), whereas Pfizer and Merck compounds yielded a negative MW correlation. Further, Pfizer compounds had an inverse relationship with TPSA, whereas compounds from Roche did not. Over the entire set of compounds, they found that the highest influence on promiscuity came from the lipophilicity and basic character of a compound.
Whilst conducting exploratory data analyses, such as that described above, it is possible that some of the descriptor correlations found will represent spurious correlations that may arise with specific data sets but are not generalizable to the end points measured. In this sense, the results are useful as hypotheses, warranting further testing over a broader range of compound classes. Still, it would be useful to determine which, if any, of these broad models are effective in prioritizing early drug candidates. It is likely that most are useful but only within a defined range of applicability. One key role of computational toxicologist is to aid in defining the applicability of in silico models to conduct analyses to understand where models can be applied most effectively and prevent the broad misconceptions that any one in silico model could be applied across all classes of chemicals.
To illustrate this point, we present a further characterization of the broad chemical subclasses of the data set used by Hughes and coworkers. 11 From Figure 2, we find that the data set, in general, has a bias towards basic drugs (56% of all compounds considered). But across all chemical classes of compounds exceeding the physicochemical thresholds found for ClogP and TPSA (compounds with ClogP > 3 and TPSA < 75), a much stronger bias is seen for lipophilic basic compounds (75% of the subclass of compounds).
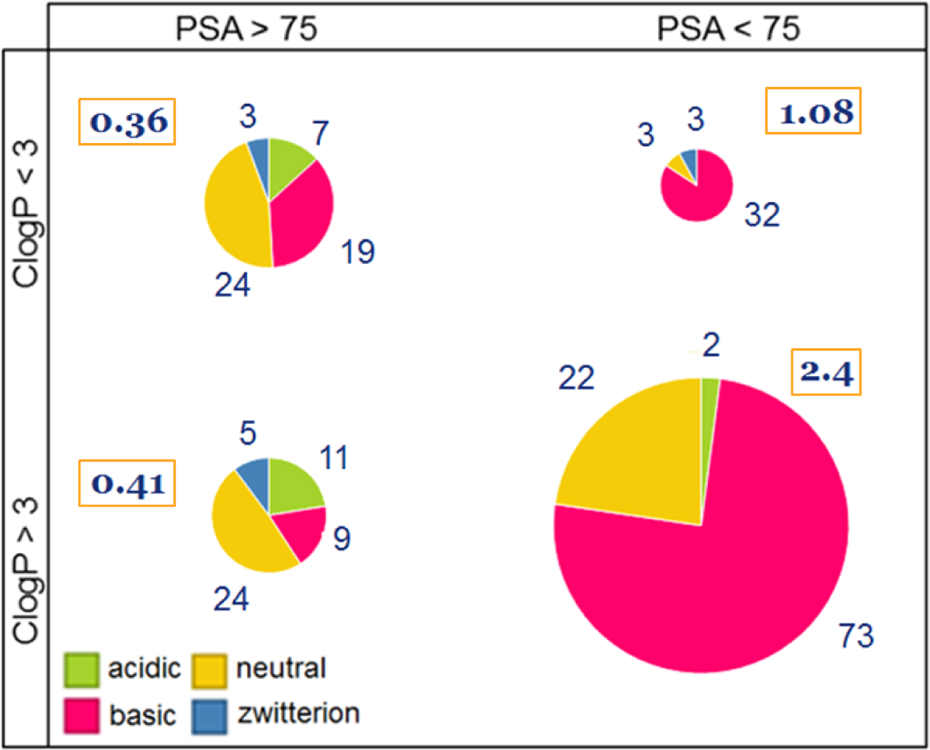
Designation of compounds in the Hughes’ in vivo data set with respect to pK a class and lipophilicity (as measured by ClogP and TPSA). Odds of toxicity from Table 1 included in orange square. TPSA: total molecular polar surface area.
Lipophilicity correlates with the ability of basic compounds to cause toxicity through general mechanisms, such as the disruption of cellular membranes, inhibition of ion channels and phospholipidosis, a lysosomal storage disorder. 14 It is possible, therefore, that the global thresholds derived from the Hughes et al. study 11 may be most applicable to basic compounds within the data set. Indeed, neutral compounds dominate the subset with TPSA > 75 (left-side pies in Figure 2), yet for this chemical class there is little difference between the likelihood of in vivo toxicity across the ClogP threshold (as shown in Table 1). This implies that the toxicity of neutral compounds within the data set, particularly those with a TPSA < 75, is not influenced by lipophilicity. This simple extra analysis into the applicability domain across chemical classes underscores the value of carefully interrogating the training data set to expose gaps in our understanding of chemical toxicology, which in this case, would be an understanding of what factors drive the toxicity of neutral compounds in the data set.
This analysis also suggests that neutral and acidic compounds must be modelled separately to explore the physicochemical properties that may be associated with in vivo toxicity, given their broadly divergent ‘absorption, distribution, metabolism, and excretion’ (ADME) properties. In extension of this awareness, any computational analysis or model should be evaluated carefully to understand the relevance of the results towards the training set and to develop a mechanistic hypothesis that can drive additional testing of the predictive performance outside of the applicability domain where knowledge gaps may silently lie.
In vitro modelling of in vivo data
Understanding the impact of the applicability domain is also essential for the development of predictive in vitro assays and in understanding where best to position them in early safety screening cascades. In this sense, the applicability domain applies to the structural and mechanistic space within the compound data set used for development and validation of the assay and how the assay data describes the in vivo end point that is being modelled.
Biochemical assays created for the idenitifcation of specific mechanistic in vivo risks, such as 5HT2b agonism for vascularopathy, 15 are useful as safety screening assays, but their utility is limited as an early predictive screen, owing to the low coverage of toxicology. In contrast, broad cellular assays, measuring cytotoxicity and mitochondrial dysfunction, are better positioned as early safety screening assays because they cover a broad range of toxicity mechanisms and can be applicable across many areas of chemical space. One disadvantage of these general assays is that their translation to in vivo end points is not straightforward or easily recognized. The ability to accurately predict in vitro–in vivo translation is important for diverting drug design into safer areas of chemical space. For early screening assays, higher accuracy can be achieved through an effective assessment of the applicability domain. The toxicological activity in an in vitro assay may not necessarily be causative of the in vivo toxicity profile being modelled and may result from a minor correlation associated with a particular chemotype or chemical class. Assays should be trained on a compound data set that covers a broad range of chemotypes and primary pharmacological mechanisms in order to reduce the chances of inference of spurious correlations, which will not be useful outside of the applicability domain of the assay. It should also be noted that the lack of an in vivo toxicity finding for a compound that shows activity in an in vitro assay may be due to exposure-related factors present in the in vivo system, such as metabolism or high clearance, which are absent in the in vitro model. It is essential, therefore, that an assay not be undervalued for potentially identifying a compound’s toxicological potential, which is mitigated in vivo. In order to evaluate an assay effectively, it is important that compounds in the training set are adequately annotated with respect to their ADME and pharmacokinetic profiles.
To prioritize the development of new assays, it is essential to identify the toxicological knowledge gaps – information that can also come from a deeper analysis of existing data. For example, studies from Shah and coworkers 16 suggest that combining the physicochemical properties of compounds with their activity in in vitro cytotoxicity assays is an effective means of identifying compounds with respect to their probability of causing adverse events in vivo. Even within this study, however, the authors point out that there are many compounds that do not follow the identified trend, and additional investigative toxicology efforts are being directed to addressing the gaps identified.
A recent area of focus is the identification of assays that describe the safety liabilities of acidic compounds. Kakiuchi-Kiyota and coworkers 17 undertook a study to understand the role of protein binding, and the impact of fetal bovine serum (FBS) levels, on results from cytotoxicity assays. In Figure 3, the results of over 70 acidic compounds screened in a cytotoxicity assay using NRK52E cells at normal (10%) or reduced (0%) concentrations of FBS is shown. It is clear from the downward shift from the diagonal of most of the data points that the reduced level of FBS enhances the apparent cytotoxicity of acidic compounds, although at different rates across the compound set. Basic and neutral compounds tested in this manner (data not shown) did not show similar cytotoxicity shifts. The effect for acidic compounds may suggest that the lack of appreciable cytotoxic activity in vitro for some compounds may well be due to poor cellular exposure. The utility of the FBS-modified cytotoxicity assay is currently being assessed across a focused data set of acidic compounds with a diversity of in vivo toxicological profiles and associated mechanisms of toxicity.
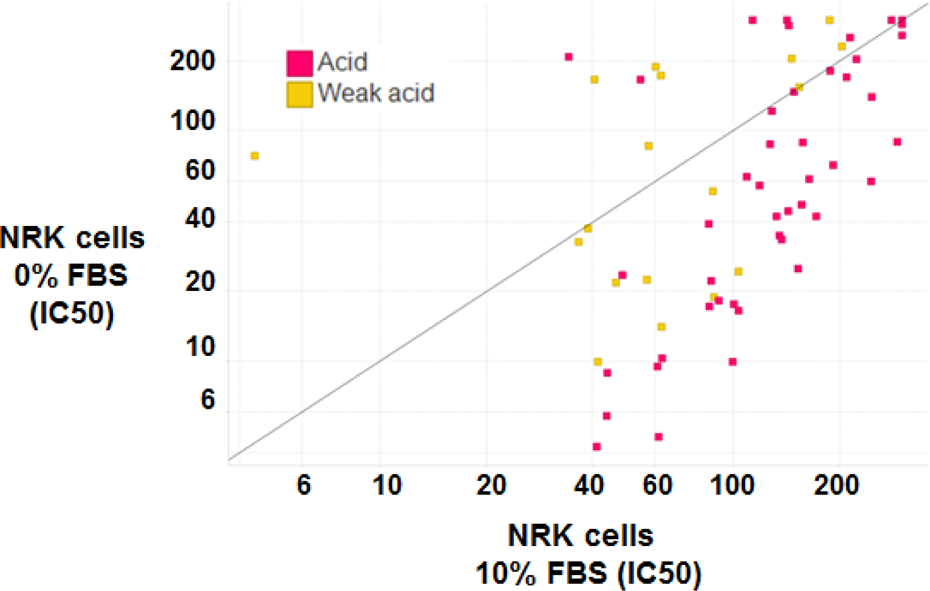
IC50 values (µM) for reduction in ATP levels for compounds tested in NRK cells in the absence and presence of FBS. IC50: half maximal inhibitory concentration; ATP: adenosine triphosphate; FBS: fetal bovine serum.
Toxicological knowledge gaps can also be addressed by a thorough and ongoing interrogation of fundamental toxicity mechanisms developed from drugs withdrawn for safety reasons. As an example, uncoupling of oxidation phosphorylation is an important mechanism of mitochondrial dysfunction that is linked to idiosyncratic organtoxicity. 18 In 2013, Naven and co-workers 19 published the results of their structure–activity relationship (SAR) studies of over 2000 compounds that were assessed in an assay to detect mitochondrial uncoupling. Through their analyses, they were able to demonstrate the importance of lipophilicity and the presence of an acidic protonophore towards promoting uncoupling activity. More importantly, however, through analyzing those compounds that did not fit the lipophilicity–protonophore trends, they were able to identify specific acidic chemotypes that were more prone to causing uncoupling activity and that should be prioritized for risk assessment early in the drug design process. They were also able to identify chemotypes that cause uncoupling through lipophilicity-independent, non-protonophoric mechanisms, such as redox cycling.
Conclusions
Computational toxicology plays a critical role in reducing late-stage attrition in drug discovery by the early prediction of a compound’s toxicological potential. If we are to improve the prediction of in vivo toxicity, however, it is urgent that we recognize the applicability and limitations of the predictive tools that frame our early screening paradigms, including both in silico models and in vitro assays.
Quantitative SAR and regression studies can be useful in identifying broad, drug design principles that reduce the likelihood of compound attrition due to general mechanisms of toxicity. Yet greater value can be achieved through identifying compounds that cause significant in vivo toxicity, despite being predicted to lie in favorable of in silico or in vitro safety space. These compounds highlight the current knowledge gaps that must be addressed if we are to improve our prediction of in vivo toxicity and avoid preventable attrition in the future.
Towards the development of new assays to address the current knowledge gaps, computational and investigative toxicologists must work together to ensure that new assays are evaluated using a focused selection of well-annotated compounds that cover a broad range of chemotypes and primary pharmacological mechanisms. This will be critical to defining the applicability domain of the assay, identifying knowledge gaps and helping provide the necessary in vitro–in vivo translation prediction for the purposes of directing drug design.
Footnotes
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.