
Abstract
Radiocontrast-induced nephropathy (RCN) is the third most common cause of acute renal failure among inpatients. Although the number of patients undergoing exams using radiocontrast is increasing, little progress has been made for RCN treatment. The pathophysiology of RCN is known as tubular injury due to oxidative stress. As autophagy regulates cellular damage under stressful conditions, we investigated the role of autophagy in RCN. RCN was induced in male C57BL/6 J mice by intraperitoneal injection of iohexol, and 3-methyladenine (3-MA) was used as an autophagy inhibitor. Tubular injury caused by iohexol was also examined in vitro using rat tubular cells (NRK-52E). Increased autophagy after iohexol administration was demonstrated by the increase of light chain 3-II in the damaged kidney tubules both in vivo and in vitro. Serum creatinine and tubular injury were significantly increased at 24 h after iohexol treatment, as compared to control group. Further they worsened with autophagy inhibition by 3-MA. In vitro studies also demonstrated that decreased cell viability by iohexol was aggravated with 3-MA pretreatment. Malondialdehyde measured for oxidative stress was increased by iohexol, and it was accentuated by autophagy inhibition, which resulted in increase of cytochrome c. Apoptosis, increased by iohexol treatment, was augmented with autophagy inhibition. Macrophage infiltration and increase of monocyte chemotactic protein-1 in kidneys were induced by iohexol, and it was aggravated with autophagy inhibition. This study showed that autophagy was involved with the pathophysiology of RCN, and the role of autophagy in modulation of apoptosis, oxidative stress, and inflammatory cell infiltration was supposed as mechanisms mitigating RCN.
Introduction
Radiocontrast-induced nephropathy (RCN) has become a growing concern due to the recent increase in number of procedures performed using contrast media. 1,2 It has become the third most common cause of acute kidney injury (AKI) in hospitalized patients. 3,4 Despite many efforts to reduce the risk of RCN, effective treatment strategies and detailed mechanisms regarding the pathophysiology of RCN remain elusive. 5,6
Autophagy, the process of degradation of damaged intracellular organelles via lysosomes, is considered to play a key role in cell survival. 7 –9 Autophagy is essential for maintaining cellular homeostasis and minimizing tissue injury against various stresses such as starvation and aging. 10,11 Previous studies have demonstrated that autophagy has cytoprotective effects in the kidneys after ischemic-reperfusion injury (IRI) as well as cisplatin or cyclosporine-induced nephropathy. 12 –18 As oxidative stress, one of the main pathogenic factors in RCN, causes derangement and disruption of intracellular structures, 19 –21 autophagy may be implicated in the pathophysiology of RCN.
In this study, we investigated the role of autophagy in renal injuries related to the use of contrast media.
Methods
Animals and models of RCN
Seven to ten-week-old male C57BL/6J mice were purchased from Orient (Seoul, Korea). All mice were housed in an animal facility with an alternating 12-h light/12-h dark cycle at 20°C and a relative humidity of 60%. All animals were acclimated for 7 days prior to all experiments. Mice were fed commercial chow and allowed free access to water until 12 h before all experiments. The Korea University Animal Care and Use Committee approved all studies (approval number: KUIACUC-2011-217).
The iodinated radiographic contrast agent used in this study was iohexol (Omnipaque, 350 mg/mL iodine and 5 g/kg iodine; GE healthcare, Princeton, New Jersey, USA). The dosages of iohexol used in the present study were determined based on the results of a preliminary study, conducted to induce consistent AKI (data are not shown). A model of RCN in mice was developed by combining an injection of a nitric oxide synthase inhibitor (NG-nitro-
Renal function and histology
All mice were killed 24 h after drug administration under ketamine anesthesia (75 mg/kg, i.p.). Blood samples were obtained from the inferior vena cava, and both kidneys were harvested after full exsanguination. Renal function was assessed by measuring serum creatinine using a Beckman analyzer II (Beckman Coulter Inc., Brea, California, USA) as described previously. 24 Tissue samples were fixed with 4% paraformaldehyde followed by paraffin embedding, and renal sections were stained with periodic acid-Schiff (PAS).
Cell culture
NRK-52E cells (renal tubular cell line of rodent origin) were obtained from American Type Culture Collection (Manassas, Virginia, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco®, Life Technologies, Seoul, Korea) and medium was replaced every 2–3 days. An in vitro model of radiocontrast injury was used as described previously. 22,23 Cells were exposed to iohexol (100 mg/mL) for 3 h. Equal volumes of saline and equiosmolar mannitol were used as the control. To examine the effect of autophagy inhibition, cells were incubated with 10 mM 3-MA with, or without, iohexol treatment.
Cell viability
As the degree of toxicity of iohexol relied upon cell viability, cell viability was measured, after treatment with saline, mannitol, and 3-MA with or without iohexol, by the 3-[4,5-dimethyl(thiazol-2-yl)-3,5-dipheryl] tetradium bromide (MTT) cytotoxic assay described previously. 22 Cells were incubated with 1 mg/mL MTT (in sterile phosphate-buffered saline) for 1 h at 37°C and then dissolved in dimethyl sulfoxide. The optical density of formazan crystals was quantified at 570 nm using a Beckman spectrophotometer.
Determination of MDA levels
Lipid peroxidation, as an index of oxidative stress, was determined by assaying malondialdehyde (MDA) production with the thiobarbituric acid reactive substance test (OxiSelect MDA adduct ELISA kit, Cell Biolabs Inc., San Diego, California, USA).
Western blot analysis
Protein was extracted from tissue samples, and protein concentration was determined using a Bradford solution (BioRad, Hercules, California, USA). To assay cytochrome c, the mitochondrial and cytosolic fractions from kidney cells were separated using a mitochondrial isolation kit, according to the manufacturer’s protocol (Pierce, Rockford, Illinois, USA). Next, 100 μg of protein were electrophoresed on sodium dodecyl sulfate–polyacrylamide gel under denaturing conditions and electroblotted to a polyvinylidene fluoride membrane (pore size: 0.45 µm; Millipore, Bedford, Massachusetts, USA). After the membrane was incubated with 5% nonfat dry milk in Tris-buffered saline Tween-20 buffer for 1 h at room temperature, the membrane was hybridized with polyclonal antibodies against light chain 3 (LC3), caspase 3, caspase 9 (dilution 1:1000; Cell Signaling Technology Inc., Beverly, Massachusetts, USA), and cytochrome c (dilution 1:1,000; BD Biosciences Clontech, San Jose, California, USA) overnight at 4°C. LC3 was measured to assess the degree of autophagy, and caspases 3 and 9 were measured to examine the degree of apoptosis. The membranes were developed with a horseradish peroxidase-conjugated secondary antibody (1:1000; Santa Cruz Biotechnology Inc., Santa Cruz, California, USA) for 90 min at room temperature. Signals were visualized by chemiluminescence detection according to the manufacturer’s protocol (Amersham Pharmacia Biotech, London, UK), and signal density measured via ImageJ (http://rsbweb.nih.gov/ij) was compared between groups. To confirm equal loading amounts of protein, β-actin antibody was used as a loading control (1:20,000; Santa Cruz Biotechnology Inc.).
Immunohistochemistry
After deparaffinization, kidney sections were hydrated in graded ethanol solutions, treated with 0.1% trypsin (Zymed, San Francisco, California, USA) and 0.3% hydrogen peroxide, and incubated with blocking serum (Vector, Peterborough, UK) to prevent nonspecific detection. Then, the slides were incubated overnight at 4°C with polyclonal LC3 antibody (1:100; Cell Signaling Technology Inc.) and F4/80 antibody to examine macrophage infiltration into mouse kidneys after iohexol treatment (1:100; AbD Serotec, Kidlington, UK). The slides were developed with a biotin-conjugated secondary antibody. For colorization, an avidin–biotin horseradish peroxidase complex (Vector Laboratories, Burlingame, California, USA) and 3,3′-diaminobenzidine tetrahydrochloride (Vector Laboratories) were applied to the slides at room temperature and counterstained with hematoxylin (Sigma-Aldrich). The negative control was stained under identical conditions with rabbit or goat serum substituted for the primary antibody. Eight to ten high-power fields (HPFs) were captured (200×; Olympus BX51, Japan), and the mean number of positive cells for F4/80 staining was calculated for quantification. Detection of apoptotic cells in the kidney was also performed on paraffin-embedded kidney tissue sections with ApopTag Plus (Intergen, Purchase, New York, USA) following the manufacturer’s protocol. Eight to ten HPFs were also captured (200×), and terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling (TUNEL)-positive cells were counted in the outer medulla and the cortex. The mean number of positive cells was calculated for quantification.
MCP-1 level measurement in renal tissue
Monocyte attractant protein-1 (MCP-1) levels of the kidneys were measured in each experimental group by enzyme-linked immunosorbent assay, according to the manufacturer’s protocol (Raybiotech, Norcross, Georgia, USA).
Statistics
Data were expressed as the mean ± standard error. Group means were compared using analysis of variance followed by Newman–Keuls post hoc analysis in GraphPad Prism version 4 (GraphPad Software, La Jolla, California, USA). Statistical significance was determined when the p value was less than 0.05.
Results
Effect of iohexol treatment on renal tubular damage and autophagy
Histologic examination with PAS staining was done with mouse kidneys from controls (Con) and iohexol-treated group (RC). Proximal tubular cell necrosis, cytoplasmic vacuolization of tubular cells, and vascular congestion were noted in the cortex and outer stripe of the medulla after iohexol treatment (Figure 1(a)). Oxidative stress, as reflected by the degree of lipid peroxidation measured by MDA assay, was significantly increased (control vs. iohexol: 1.178 ± 0.287 vs. 2.947 ± 0.759 μM MDA/g/mL protein, p < 0.001; Figure 1(b)). When autophagy was examined by LC3 expression, increased LC3 II, converted from LC3 I, was seen after iohexol treatment in NRK-52E cells in comparison to saline treatment (Figure 2(a), p < 0.05). Immunohistochemistry of LC3 in mouse kidneys, after iohexol administration, also revealed increased LC3 expression in damaged tubules when compared to the saline treatment group (Figure 2(b)).

Iohexol treatment induced renal tubular injury and increased oxidative stress. (a) Histologic examination with PAS staining of mice kidneys showed proximal tubular necrosis and cytoplasmic vacuolization after iohexol treatment compared to controls (×200). (b) Oxidative stress measured by MDA assay was significantly increased. *p < 0.05: compared to Con. Con: saline-treated group; RC: iohexol-treated group; PAS: periodic acid-Schiff; MDA: malondialdehyde.
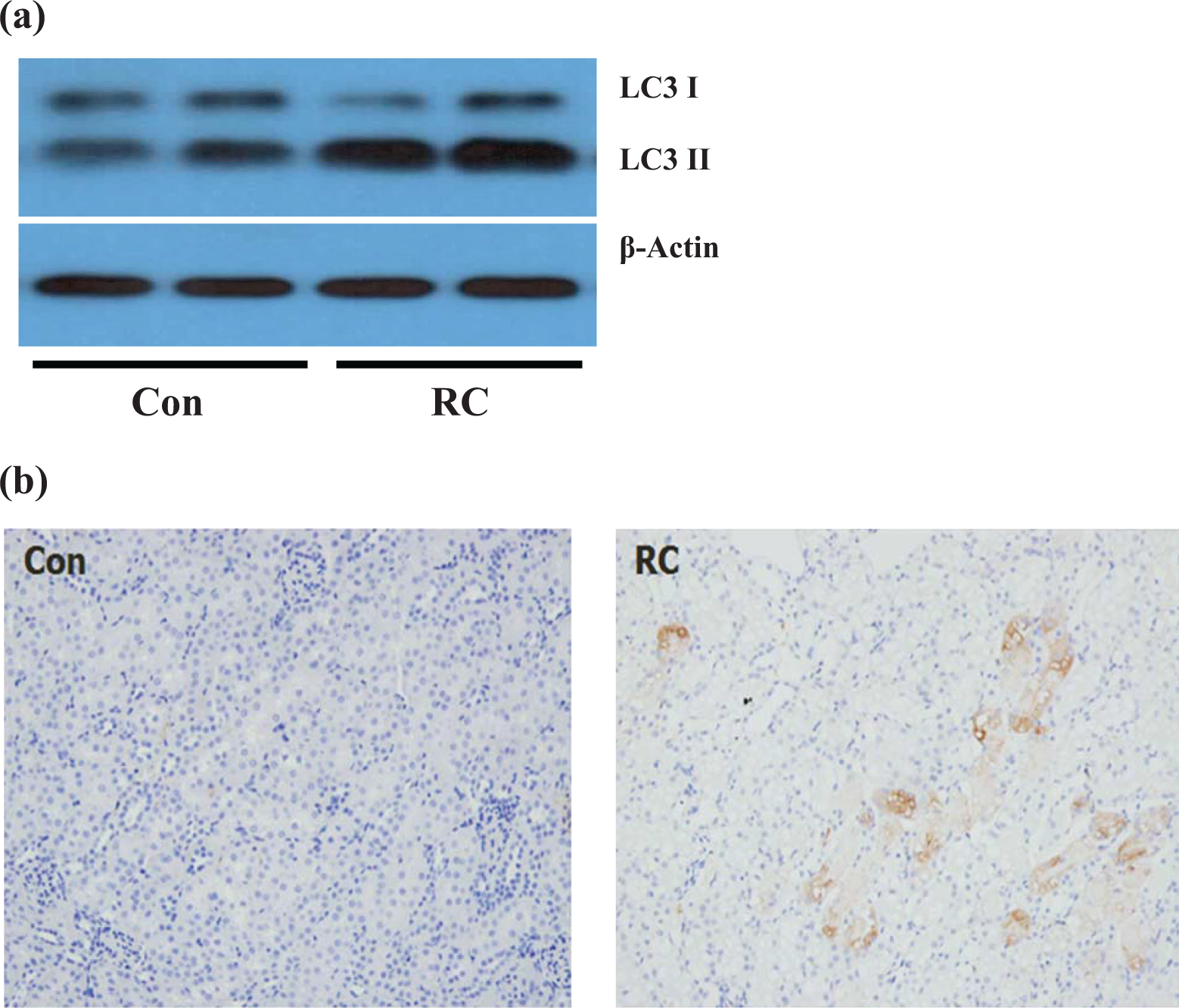
(a) Autophagy was induced in renal tubules after iohexol treatment. Iohexol treatment led the conversion of LC3 I to LC3 II, and so increase of LC3 II was demonstrated after iohexol treatment group compared to controls. (b) Immunohistochemistry of LC3 in mice kidney also revealed LC3 accumulation in damaged tubules after iohexol administration. Con: saline-treated group; RC: iohexol-treated group; LC3: light chain 3.
Effect of autophagy inhibition on renal dysfunction and tubular damage after iohexol treatment
To examine the role of autophagy in RCN, we used an autophagy inhibitor, 3-MA, which inhibited autophagy significantly, as measured by LC3 II expression, converted from LC3 I, in the kidney (data are not shown). Serum creatinine increased significantly 24 h after iohexol administration compared to control, saline treatment group. Mannitol, which was used as a control for examining the effect of high osmolarity of iohexol, did not induce changes in serum creatinine levels. It could be postulated that renal injury, after iohexol treatment, was not simply caused by changes in the osmolarity resulting from iohexol administration. Autophagy inhibition after iohexol administration elevated serum creatinine to a greater extent compared to other groups, such as saline, mannitol, 3-MA, and iohexol-only treatment group, which were mentioned above (control, 0.27 ± 0.04 mg/dL; mannitol, 0.24 ± 0.03 mg/dL; 3-MA, 0.36 ± 0.05 mg/dL; iohexol, 0.91 ± 0.5305 mg/dL; iohexol + 3-MA, 1.31 ± 0.2405 mg/dL; p < 0.05; Figure 3(a)). Upon histological examination, tubular damages noted after iohexol treatment (Figure 3(e)) were not observed in other groups including saline, mannitol, and 3-MA-only treatment group (Figure 3(b) to (d)). Additionally, tubular vacuolization and necrosis worsened with autophagy inhibition after 3-MA treatment (Figure 3(f)).

Autophagy inhibition with 3-MA aggravated iohexol-induced acute kidney injury in murine model. (a) Serum creatinine was elevated at 24 h after iohexol treatment, and it was accentuated with autophagy inhibition by 3-MA (n = 10). (b) Con. (c) Man. (d) 3-MA. (e) Iohexol treatment induced proximal tubular necrosis and cytoplasmic vacuolization, and (f) it was worsen by 3-MA (PAS, ×200). *p < 0.05: compared to Con, Man, and 3-MA groups; # p < 0.05: compared to RC group. Con: saline-treated group; Man: mannitol-treated group; 3-MA: 3-methyladenine-only treated group; RC: iohexol-treated group; RC + 3-MA: iohexol and 3-methyladenine-treated group; PAS: periodic acid-Schiff.
Effect of autophagy inhibition on viability of renal tubular cells
Iohexol induced significant tubular cell injury, and cell viability measured by MTT assay was significantly reduced after iohexol treatment. Autophagy inhibition with 3-MA pretreatment aggravated the viability of tubular cells after iohexol treatment (control, 1.191 ± 0.257 mg/dL; mannitol, 1.071 ± 0.067 mg/dL; 3-MA, 0.980 ± 0.080 mg/dL; iohexol, 0.395 ± 0.040 mg/dL; iohexol + 3-MA, 0.290 ± 0.036 mg/dL; p < 0.05; Figure 4).

Cell viability was measured by MTT cytotoxic assay. It was decreased after iohexol treatment in rat tubular cells and aggravated with autophagy inhibition by 3-MA. *p < 0.05: compared to Con, Man, and 3-MA groups; # p < 0.05: compared to RC group. Con: saline-treated group; Man: mannitol-treated group; 3-MA: 3-methyladenine-only treated group; RC: iohexol-treated group; RC + 3-MA: iohexol and 3-methyladenine-treated group; MTT: 3-[4,5-dimethyl(thiazol-2-yl)-3,5-dipheryl] tetradium bromide.
Effect of autophagy inhibition on apoptosis
Saline, mannitol, and 3-MA-only treatment did not increase the number of brown-colored TUNEL-positive cells indicating apoptosis (Figure 5(a) to (c)). Increased apoptosis in renal tubules of mice was manifested at 24 h after iohexol treatment via TUNEL staining (Figure 5(d)). The number of apoptotic cells, after iohexol treatment, was more increased by autophagy inhibition with 3-MA administration (Figure 5(e)). The differences were statistically significant when compared to the other groups such as saline, mannitol, 3-MA, and iohexol-only treatment groups (control, 0.30 ± 0.15/HPFs; mannitol, 0.60 ± 0.22/HPFs; 3-MA, 0.60 ± 0.22/HPFs; iohexol, 16.90 ± 4.28/HPFs; iohexol + 3-MA, 31.30 ± 2.65/HPFs; p < 0.001; Figure 5(f)). Immunoblot analysis of caspase 3 in renal tubular cells showed similar results. Cleaved caspase 3 expression in tubular cells was significantly increased after iohexol treatment, which was accentuated by autophagy inhibition with 3-MA (Figure 6(a) and (b), p < 0.01).

(a) Con. (b) Man. (c) 3-MA. (d) Iohexol treatment induced the increase of apoptotic cells (dark brown color) in TUNEL staining, and it was significantly aggravated with autophagy inhibition by 3-MA (e and f; TUNEL, ×400). Con: saline-treated group; Man: mannitol-treated group; 3-MA: 3-methyladenine-only treated group; RC: iohexol-treated group, RC + 3-MA: iohexol and 3-methyladenine-treated group; *p < 0.05: compared to Con, Man, and 3-MA groups; # p < 0.05: compared to RC group; TUNEL: terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling.

(a and b) Immunoblotting of caspase 3 showed that iohexol treatment induced apoptosis, the increase of caspase 3 expression, in rat tubular cells, and it was significantly aggravated with autophagy inhibition by 3-MA. *p < 0.05: compared to Con, Man, and 3-MA groups; # p < 0.05: compared to RC group. Con: saline-treated group; Man: mannitol-treated group; 3-MA: 3-methyladenine-only treated group; RC: iohexol-treated group; RC + 3-MA: iohexol and 3-methyladenine-treated group.
Effect of autophagy inhibition on mitochondrial injury
After iohexol treatment, increased cytochrome c expression in the cytosol was documented by in vivo and in vitro experiments. Autophagy inhibition, by 3-MA treatment, accentuated cytochrome c release in both in vivo (Figure 7(a) and 7(b), p < 0.001) and in vitro experiments (Figure 7(b), 7(c) and 7(d), p < 0.05). Iohexol treatment induced caspase 9 activation, which was also augmented by autophagy inhibition with 3-MA pretreatment (Figure 7(c), 7(e) and 7(f), p < 0.001).

Immunoblotting examinations demonstrated that iohexol treatment induced cytochrome c release into cytosol in vivo and in vitro experiments (a and c), and increased caspase 9 expression (e). Those changes were significantly accentuated with autophagy inhibition by 3-MA (b, d, and f). *p < 0.05: compared to Con, Man, and 3-MA groups; # p < 0.05: compared to RC group. Con: saline-treated group; Man: mannitol-treated group; 3-MA: 3-methyladenine-only treated group; RC: iohexol-treated group; RC + 3-MA: iohexol and 3-methyladenine treated group.
Effect of autophagy inhibition on macrophage infiltration
Aside from the iohexol-treated group, saline, mannitol, and 3-MA-only treatment groups did not show macrophage infiltration in F4/80 staining (Figure 8(a) to (c)). The number of F4/80 positive cells in mice kidneys increased after iohexol treatment (Figure 8(d)). Autophagy inhibition with 3-MA administration resulted in significant increases in the number of macrophages of kidneys following iohexol treatment (Figure 8(e)), and this increase was statistically significant compared to other groups (control, 0.80 ± 0.13/HPFs; mannitol, 0.50 ± 0.16/HPFs; 3-MA, 0.50 ± 0.16/HPFs; iohexol, 9.64 ± 0.48/HPFs; iohexol + 3-MA, 16.35 ± 1.23/HPFs; p < 0.001, Figure 8(f))

(a) Con. (b) Man. (c) 3-MA. (d) Iohexol treatment induced F4/80(+) macrophage infiltration (red arrows) into renal tubules in mice kidneys, and it was significantly aggravated with autophagy inhibition by 3-MA (e and f; F4/80, ×400). *p < 0.05: compared to Con, Man, and 3-MA groups; # p < 0.05: compared to RC group. Con: saline-treated group; Man: mannitol-treated group; 3-MA: 3-methyladenine-only-treated group; RC: iohexol-treated group; RC + 3-MA: iohexol and 3-methyladenine-treated group.
Effect of autophagy inhibition on chemokine expression
MCP-1, a chemokine related to macrophage activation, was increased after iohexol treatment compared to other groups such as saline, mannitol, and 3-MA-only treatment group. Autophagy inhibition, with 3-MA, augmented the increase of MCP-1 in kidneys (control, 21.6 ± 0.81; mannitol, 21.1 ± 0.63 pg/mL; 3-MA, 22.3 ± 0.84 pg/mL; iohexol, 35.6 ± 3.31 pg/mL; iohexol + 3-MA, 47.1 ± 3.53 pg/mL; p < 0.001; Figure 9).

MCP-1 expression in mice kidneys significantly increased after iohexol treatment, and it was significantly accentuated with autophagy inhibition by 3-MA. *p < 0.05 compared to Con, Man, and 3-MA groups; # p < 0.05: compared to RC group. Con: saline-treated group; Man: mannitol-treated group; 3-MA: 3-methyladenine-only-treated group; RC: iohexol-treated group; RC + 3-MA: iohexol and 3-methyladenine-treated group; MCP-1: monocyte attractant protein-1.
Discussion
In this study, we demonstrated that autophagy contributes to renoprotection in RCN, and this finding was mediated by the modulation of apoptosis and mitochondrial injury.
RCN, defined as a decline in renal function of more than 25%, or 0.5 mg/dL increase in creatinine 24–72 h after the use of contrast media, 1,25 is a common cause of AKI and accounts for 2–20% of AKI in hospitalized patients. 2,3,26 In addition to increasing costs due to prolonged hospitalization, there are also substantial increases of morbidity and mortality: the 1-year mortality rate increases by up to 30%. 6,27 RCN also increases the risk of irreversible deterioration of renal function as a part of the progression to end-stage renal disease. 28 The prevalence of RCN is increasing due to the rapid growth of the population at high risk for RCN, such as elderly, diabetic patients, and those with chronic kidney disease, as well as increases in the number of diagnostic procedures and therapeutic interventions, which requires the use of contrast media. 6 Despite advances in developing safer contrast media 29 –31 and various preventive measures such as hydration and prophylaxis with antioxidants such as N-acetylcysteine, 4,32,33 no definitive progress has been made toward decreasing the risk of RCN yet. Hemodynamic changes, such as decreases in renal blood flow and oxidative stress related to the generation of reactive oxygen species, may be factors related to the development of RCN. 26,34 –36 However, the exact pathophysiology of RCN is not clear yet.
Autophagy is an intracellular process that degrades and recycles damaged organelles and aggregated proteins to sustain cellular homeostasis. 7 –9 Although autophagy is known to be triggered as a cellular reaction to physiological changes such as nutrient depletion and starvation, 37 recent studies have also demonstrated that autophagy is closely associated with the pathogenesis of various renal tubular injuries, such as IRI, cisplatin- or cyclosporine-induced nephrotoxicity. Therefore, renal protective roles of autophagy have been suggested previously. 12 –18 In the present study, we found that the degree of autophagy, measured by the expression of LC3 II, was very low in the kidney under normal conditions, and it was significantly increased in damaged tubules after treatment with iohexol. This finding indicates that autophagy is involved in tubular injury caused by iohexol. LC3 II is an autophagosome membrane-bound protein converted from a cytosolic protein, LC3 I. Conversion of LC3 I to LC3 II is one of the reliable markers for autophagy. Increased oxidative stress, measured by MDA assay, also accompanied with tubular damage and increased autophagy after iohexol treatment. We hypothesized that autophagy might be induced as an adaptive process for cell survival in RCN, although it is not clear whether autophagy itself provoked cell death or was passively increased for cellular defense. To confirm the role of autophagy and determine whether it is protective or detrimental in RCN, pharmacological inhibition of autophagy, using 3-MA, was utilized. 38 Inhibition of autophagy resulted in significant aggravation of functional and histologic renal damage in vivo experiment, as well as tubular cell viability, measured by MTT assay, and in vitro experiments. These findings indicate that autophagy exerted a renoprotective effect on RCN in a murine model. In previous studies of AKI, the exact role of autophagy on protection from AKI has remained controversial. Proper activation of autophagy was needed to decrease tubular injury by ischemia/reperfusion, cisplatin, or cyclosporine. 12 –18 In contrast, some previous studies reported that increased autophagy resulted in aggravation of tubular cell death in IRI and tunicamycin-induced renal injury. 39,40 The effect of autophagy on renal injury may depend on the specific conditions and individual characteristics of stress factors; in this study, autophagy was found to be beneficial for the prevention of RCN.
The detailed mechanisms of how autophagy protects the kidney against various stresses are much less clear. In the RCN model of the present study, increases of cytochrome c in the cytosol, after iohexol treatment, were demonstrated both in vivo and in vitro experiments. It is presumably suggested that oxidative stress, increased by iohexol in renal tubules, may cause mitochondrial membrane potential loss and lead to the release of cytochrome c from mitochondria into the cytosol. Cellular stress, related to oxidative stress and mitochondrial depolarization, was thought to be closely related to autophagy. 41 Because autophagy is known to eliminate damaged organelles, which are potentially cytotoxic for cell viability, inhibition of autophagy may accelerate mitochondrial depolarization and cellular damage. In the present study, increased cytochrome c by iohexol treatment in cytosol was augmented after autophagy inhibition. Increases of cytochrome c, as a result of iohexol treatment, triggered caspase-cascade activation, initially shown as an increase of caspase 9. Well-known initiator of the caspase-cascade, caspase 9, which was activated by mitochondrial injury, led to the activation of caspase 3, and subsequently resulted in apoptosis of the proximal tubules. Apoptosis is known as a mechanism involved in renal tubular injury in various conditions including RCN, 22,23,42 and the interaction between autophagy and apoptosis, as mechanisms controlling cell survival, was suggested previously. 18,43 Under the circumstances that autophagy did not function properly for maintaining cellular homeostasis against the oxidative stress induced by iohexol, apoptosis of renal tubules increased, and it may result in further tubular damage. Mitochondrial depolarization and apoptosis induced by iohexol led to macrophage infiltration into the kidney measured by F4/80 staining. Elevated expression of MCP-1, a cytokine related to macrophage activation, was also observed. Inflammatory cell infiltration and increases of inflammatory cytokine were accentuated by autophagy inhibition. Previous studies also indicated that inflammatory cells and cytokines were involved in the pathogenesis of RCN, 44,45 and inflammatory process may be one of the mechanisms mediating RCN through autophagy, as shown in this study.
Although the association of autophagy with RCN has not been reported previously, this study has several limitations. First, immunoblots revealing the conversion of LC3 I to LC3 II was the only method used to demonstrate autophagy in renal tubules after iohexol administration. To detect autophagy, visualization of the autophagosome via electron microscopy or green fluorescent protein-LC3 emergence after plasmid transfection by immunofluorescence microscopy is often used in combination with immunoblot assays. 14 Due to a technical problem, only immunoblot studies were adapted in the present study. However, immunoblot assays have been considered as a reliable method to check autophagy in many studies. 14,15,17 Moreover, immunohistochemistry with LC3 also revealed that LC3 appeared in damaged tubules after iohexol injury, which was another evidence supporting the hypothesis that autophagy was related to tubular damage by iohexol. Second, only pharmacologic inhibition was adapted to examine the role of autophagy. In our laboratory, genetically engineered animals defective in the autophagy process were not available. Pharmacologic activators of autophagy, such as an mammalion target of rapamycin (mTOR) inhibitor, were also used to check the role the present study because mTOR inhibition does not selectively boost autophagy but also activates various cellular pathways leading to bias.
In conclusion, increased autophagy was demonstrated in the kidneys after the administration of contrast media, and inhibition of autophagy worsened functional and histologic renal dysfunction in RCN, which suggested the protective role of autophagy in the setting of RCN. Autophagy was associated with the pathophysiology of RCN, as a cellular process to cope with stressful conditions, increased apoptosis and inflammation cell. In the future, studies to facilitate autophagy in order to reduce cellular stress inflicted via contrast media may be helpful for the development of new therapeutic strategies for the treatment of RCN.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant for young investigators from the Korean Society of Nephrology 2013 and by a grant from Korea University (NARS3270Q).