
Abstract
Background:
Perfluorooctanoic acid (PFOA) is one of the most widely used perfluoroalkanes as surfactants, lubricants and processing aids in the production of polymers, which has also been detected in the environment, wildlife and human body. Animal studies indicated that PFOA caused a wide array of toxic effects including liver and brain dysfunction, carcinogenicity and reproductive and developmental toxicity. Based on the established role of mitochondria-mediated pathways in the observed toxic effects of many drugs and chemicals, in this study, the potential toxic effects of PFOA on mitochondria isolated from rat liver and brain have been investigated.
Method:
Mitochondria were isolated by differential centrifugation method and incubated with different concentrations of PFOA (0.5–1.5 mM). The effects of PFOA were assessed on a series of mitochondrial parameters including reactive oxygen species (ROS) formation, activities of mitochondrial complexes I/II/III, reduced glutathione (GSH) content, adenosine triphosphate (ATP) level, membrane potential, lipid peroxidation (LPO), mitochondrial swelling and cytochrome c release.
Results:
The data on liver mitochondria indicated that PFOA-induced ROS elevation in both mitochondrial complexes I and III, mitochondrial membrane potential collapse, swelling, cytochrome c release and decreased ATP level which induces apoptosis or necrosis. On brain mitochondria, PFOA showed fairly similar effects on the above-mentioned parameters. However, different results were obtained when the effect of PFOA was assessed on LPO and complex II activity.
Conclusions:
Due to the fact that PFOA had toxic effects on the mitochondria isolated, it could be suggested that mitochondrial toxicity could be a plausible mechanism for the toxic effects of this fluorochemical on liver and brain function.
Introduction
Perfluoroalkanes have long been used as surfactants, lubricants and processing aids in the industrial applications such as Teflon production and food packaging materials. 1,2 Among them, perfluorooctanoic acid (PFOA) is the dominant perfluoroalkyl acid detected in the environment, wildlife and human. 3,4 Human exposure to PFOA is due to the widespread environmental persistence and the high usage of it in food industries. 4 Animal studies have shown that exposure to PFOA is contributed to the vast majority of outcomes including hepatotoxicity, carcinogenicity, immunotoxicity and developmental disturbances. 2,5 Regarding the pivotal role of liver in metabolism and clearance of xenobiotics, exposure to PFOA is associated with macroscopic and microscopic changes in hepatocytes, 6 hepatocellular adenoma 7 and increased amino tranferase activity. 8 It has been reported that hepatocellular toxicity of PFAO is partly related to induction of apoptosis via boosting the oxidative stress generation. 2
Mitochondria not only produce large levels of energy in highly metabolic organ but also are contributed to the regulation of physiological and pathophysiological conditions. Brain and liver are highly sensitive tissues to decrease in energy metabolism. 9 Large bodies of evidence have demonstrated the key role of mitochondrial function in the vast majority of disorders related to the brain and liver. 9 Therefore, mitochondria have key role as target organelle for toxicity mechanisms of xenobiotics such as heavy metals and chemicals such as PFOA. 5,10,11 In addition, other studies reported that perfluoroalkyl acids are able to induce alterations in mitochondrial performance such as respiration and structural stability in both hepatocytes (HepG2) and cerebellar granule cells via induction of mitochondrial MPT and inhibition of alteration of membrane stability. 11 – 13 Based on the above discussions and importance of mitochondria-mediated pathways in the toxicity of different chemicals, 14,15 we evaluated the effects of PFOA on the biochemical functions of mitochondria isolated from rat liver and brain tissue.
Materials and methods
Materials
All the chemicals and reagents were purchased from Sigma-Aldrich (Taufkirchen, Germany) with the best analytical grade.
Animals
Male Sprague Dawley rats were purchased from Pasteur Institute, Tehran, Iran. Animals were fed with standard chow diet and water ad libitum. The animal experimentation protocols had been approved by the Animal Ethics Committee of Zanjan University of Medical Sciences. The ethical standards were based on European Convention for the Protection of Vertebrate Animals Used for Experimental and other Scientific Purposes Acts of 1986, and the Guiding Principles in the Use of Animals in Toxicology, adopted by the Society of Toxicology in 1989, for the acceptable use of experimental animals. All the animals used for the study received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86–23, revised 1985).
Isolation of mitochondria
Animals were decapitated and their liver and brain were dissected out, which were rapidly used for the experiments. Mitochondria were isolated using ultracentrifugation method described in a previous report. 16 Mitochondrial protein concentration was determined by Bradford method with bovine serum albumin as a standard and adjusted to 500 µg protein/ml in all the experiments. 17
Quantification of mitochondrial ROS
Flow cytometry
Mitochondrial suspensions were incubated with 0.5–1.5 mM concentrations of PFOA in respiration buffer containing 0.32 mM sucrose, 10 mM Tris-hydrochloric acid, 20 mM 3-(N-morpholino)propanesulfonic acid, 50 μM ethylene glycol tetraacetic acid, 0.5 mM magnesium chloride, 0.1 mM potassium dihydrogen phosphate and 5 mM sodium succinate. 18 After the addition of PFOA, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) was added (final concentration of 10 μM) to the mitochondrial suspension and incubated for 20 min. PFOA-induced ROS generation in isolated mitochondria was measured using a flowcytometer (Partec, Deutschland) equipped with a 488 nm argon ion laser and supplied with the Flomax software, and the signals were obtained using a 530 nm bandpass filter (FL-1 channel). Each determination was based on the mean fluorescence intensity of 15,000 counts.
Fluorimetry
Mitochondrial ROS level was also measured using DCFH-DA reagent using a Hitachi fluorescence spectrophotometer (Japan) according to our previous work. 19
Complex I/III activity assay
The effect of PFOA on the activity of complexes I and III was evaluated through the spectrophotofluorometric measurement of ROS production using homovanillic acid and horseradish peroxidase. ROS formation in complex I was measured in the presence of pyruvate 2.5 mM + malate 2.5 mM (p/m), p/m + PFOA (0.5 mM) and p/m + PFOA (0.5 mM) + rotenone 2 µM (p/m + PFOA + Rot). ROS formation in complex III was measured in the presence of succinate 5 mM (Suc), Suc + PFOA (0.5 mM) and Suc + PFOA (0.5 mM) + antimycin A 2 µM (Suc + PFOA + AA). In both assays, ROS formation was measured 30 min after the treatments of isolated rat mitochondria with the reagents mentioned above. The excitation and emission wavelengths were set at 312 and 420 nm, respectively.
Assessment of complex II activity
The activity of mitochondrial succinate dehydrogenase (complex II) was evaluated by measuring the reduction of MTT to formazan with an enzyme-linked immunosorbent assay (ELISA) reader (InfiniteM 200, TECAN, Switzerland) set at 570 nm. 20
LPO assay
Malondialdehyde (MDA) content was determined by measuring the absorbance of the test samples at 532 nm using an ELISA reader (InfiniteM 200, TECAN). 21
Determination of GSH content
Mitochondrial GSH content was assayed using 5,5′-dithiobis-(2-nitrobenzoic acid) at 412 nm with an ELISA reader (InfiniteM 200, TECAN). GSH content was expressed as microgram per milligram protein. 22
MMP assay
Mitochondrial membrane potential (MMP) was measured fluorometrically using rhodamine123 as the fluorescent probe at excitation and emission wavelengths of 490 nm and 535 nm, respectively. 23
Determination of ATP level
The effect of PFOA on mitochondrial adenosine triphosphate (ATP) level was measured using luciferase enzyme. 24 Bioluminescence intensity was measured using a Sirius tube luminometer (Berthold Detection System, Germany).
Determination of mitochondrial swelling
Mitochondrial swelling was measured by the absorbance change in mitochondrial suspensions recorded at 540 nm using ELISA reader following the procedure described by Zhao et al. 20
Cytochrome c assay
The amount of released cytochrome c was assayed in a microplate reader at 450 nm using the Quantikine® rat/mouse cytochrome c immunoassay kit (Minneapolis, Minnesota, USA). The assay was performed according to the instructions provided by the manufacturer.
Statistical analysis
Results have been presented as mean ± SEM. All statistical analyses were performed using Statistical Package for the Social Sciences Version 17 software. Assays were performed in triplicate and the mean was used for the statistical analyses. Statistical significance was determined using the one-way analysis of variance (ANOVA) test, followed by the post hoc Tukey’s test. In some experiments, the two-way ANOVA test, followed by the post hoc Bonferroni test was also performed. The value of p < 0.05 was considered to be statistically significant.
Results
Effect of PFOA on mitochondrial ROS production
Treatment of liver mitochondria with PFOA (0.75 to 1.5 mM) resulted in significant elevation of ROS (p < 0.05, Tables 1 and 2, Figure 1). Similar results were observed in brain mitochondria where significant PFOA-induced elevation of ROS was observed from 0.5 mM to 1.5 mM (p < 0.05). Notably, elevation of ROS in both liver and brain mitochondria was time and concentration dependent (Tables 1 and 2, Figure 1).
PFOA-induced ROS formation in isolated liver and brain mitochondria.a

PFOA: perfluorooctanoic acid; ROS: reactive oxygen species; DCFH-DA: 2′,7′-dichlorodihydrofluorescein diacetate.
aROS formation was determined by flow cytometry using DCFH-DA as described in Materials and methods and demonstrated as fluorescence intensity of DCF. Values are represented as mean ± SEM (n = 3). Control group contains respiration buffer plus DCFH-DA (10 µM).
bFluorescence intensity (%) is described as the percentage of fluorescence intensity compared with control mitochondria.
c p < 0.05: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA) at the same time interval.
d p < 0.001: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA) at the same time interval.
PFOA-induced ROS formation in isolated mitochondria.a

PFOA: perfluorooctanoic acid; ROS: reactive oxygen species; DCFH-DA: 2′,7′-dichlorodihydrofluorescein diacetate.
aROS formation was determined by spectroflorimetrically using DCFH-DA as described in Materials and methods. The fluorescence intensity was recorded after addition of various concentrations of PFOA (0.5, 0.75, 1 and 1.5 mM) at time intervals 5–60 min. Values represented as mean ± SEM (n = 3).
bCalculated based on the formula:
c p < 0.05: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA) at the same time interval.
d p < 0.01: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA) at the same time interval.
e p < 0.001: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA) at the same time interval.
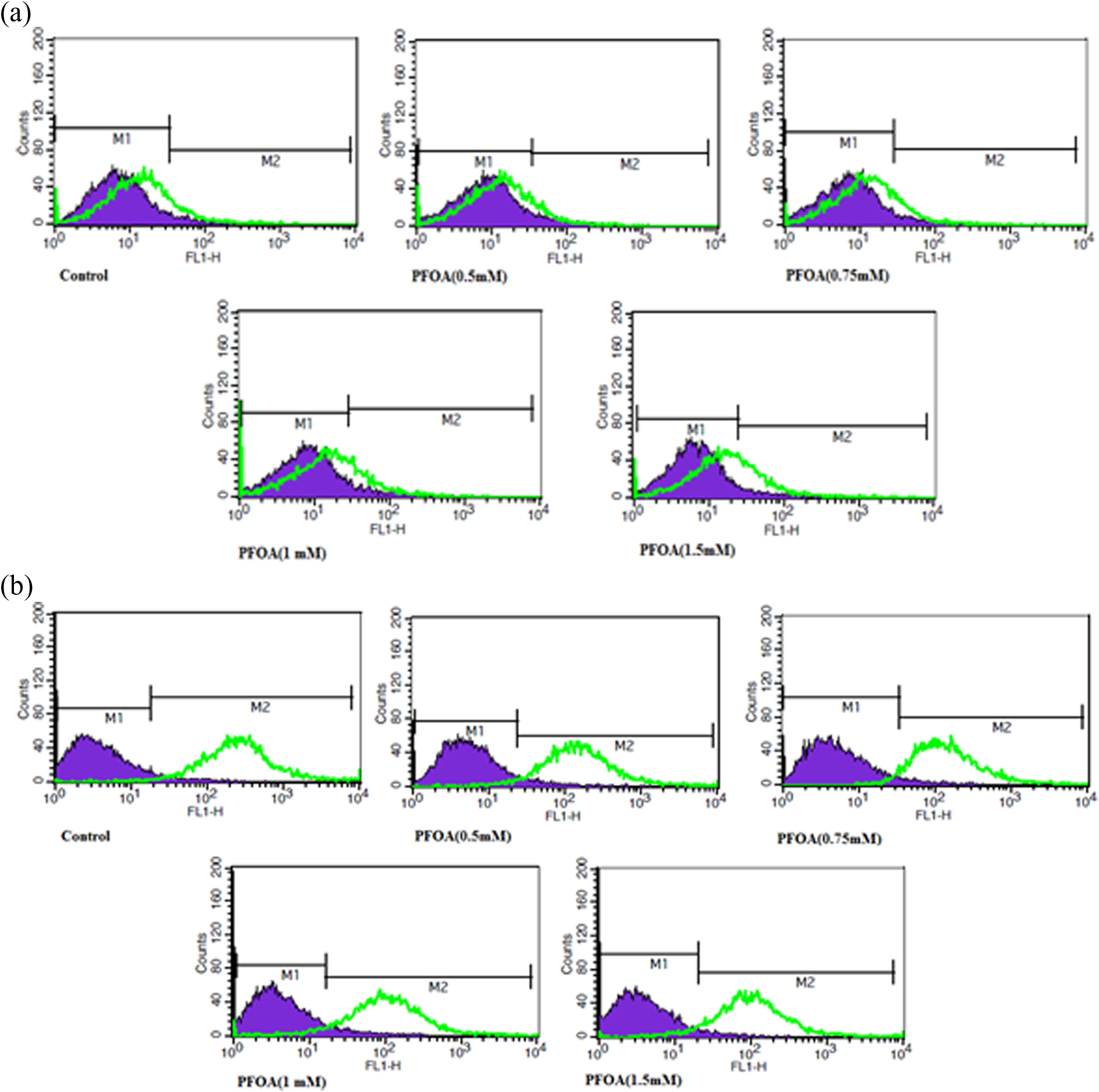
Flowcytograms obtained through the evaluation of ROS formation in isolated liver (a) and brain (b) mitochondria. ROS formation was determined by flow cytometry using DCFH-DA as described in materials and methods and demonstrated as fluorescence intensity of DCF. Control group contained respiratory buffer plus DCFH-DA (10 µM). ROS: reactive oxygen species; DCFH-DA: 2′,7′-dichlorodihydrofluorescein diacetate.
Effect of PFOA on complex I/III activity
Based on our data, incubation of liver and brain mitochondria with 0.5 mM PFOA significantly increased ROS production in both complexes I and III in similar fashion (p < 0.05, Table 3).
Influence of respiratory substrates and inhibitors of complexes I and III on PFOA-induced ROS formation in isolated mitochondria.a
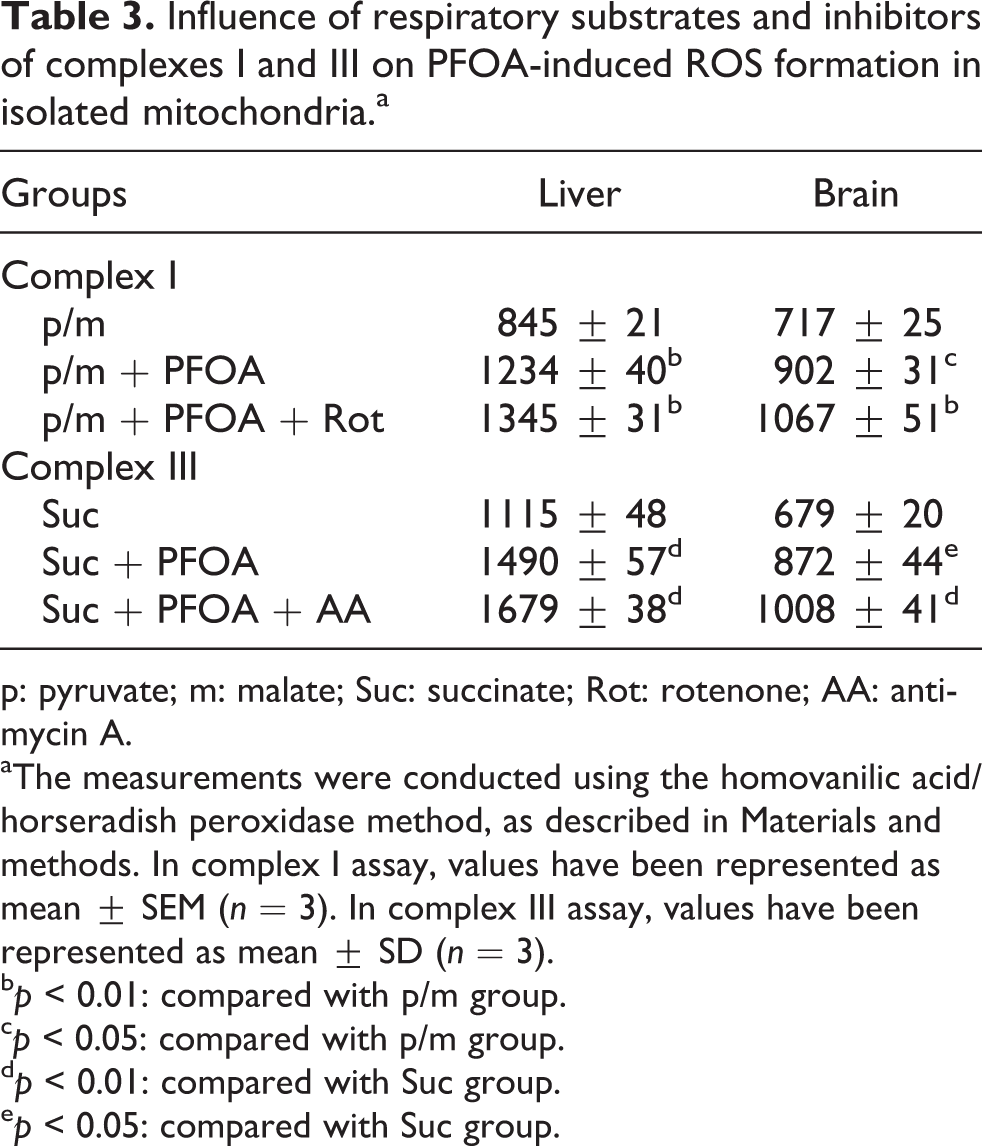
p: pyruvate; m: malate; Suc: succinate; Rot: rotenone; AA: antimycin A.
aThe measurements were conducted using the homovanilic acid/horseradish peroxidase method, as described in Materials and methods. In complex I assay, values have been represented as mean ± SEM (n = 3). In complex III assay, values have been represented as mean ± SD (n = 3).
b p < 0.01: compared with p/m group.
c p < 0.05: compared with p/m group.
d p < 0.01: compared with Suc group.
e p < 0.05: compared with Suc group.
Effect of PFOA on mitochondrial succinate dehydrogenase (complex II) activity
PFOA significantly inhibited complex II activity of liver mitochondria in a concentration-dependent manner from 0.75 mM to 1.5 mM (p < 0.05, Figure 2). In case of brain mitochondria, complex II activity was affected by only the highest concentration of PFOA (1.5 mM, Figure 2). Based on our results the order of sensitivity of liver against PFOA toxicity is higher than brain tissue.

Effect of PFOA on succinate dehydrogenase (complex II) activity. Succinate dehydrogenase activity was measured using MTT dye as described in Materials and methods. Liver and brain mitochondria (0.5 mg/ml) were incubated at 30°C for 1 h with various concentrations of PFOA (0.5, 0.75, 1, 1.5 mM). Values are represented as mean ± SEM (n = 3). *p < 0.05; ***p < 0.001: compared with control mitochondria. PFOA: perfluorooctanoic acid; MTT: 3-(4,5-dimethylthiazol-2-yl)-12,5-diphenyltetrazolium bromide.
Effect of PFOA on mitochondrial LPO
Exposure of liver mitochondria to PFOA did not show any significant increase in MDA content as the marker of LPO (Figure 3). On the contrary, PFOA (0.5–1.5 mM) significantly increased LPO in brain mitochondria in a concentration-dependent manner (p < 0.05, Figure 3) due to relationship between the concentration of polyunsaturated fatty acids and susceptibility to LPO.

Effect of PFOA on mitochondrial LPO. Liver and brain mitochondria (0.5 mg/ml) were incubated at 30°C for 1 h with PFOA (0.5, 0.75, 1, 1.5 mM). Values are presented as mean ± SEM (n = 3). **p < 0.01; ***p < 0.001: compared with control mitochondria. PFOA: perfluorooctanoic acid; LPO: lipid peroxidation.
Effect of PFOA on mitochondrial GSH content
Treatment of mitochondria with different concentrations of PFOA did not lead to any significant alteration in GSH content in a similar manner on isolated rat liver and brain mitochondria (Figure 4).
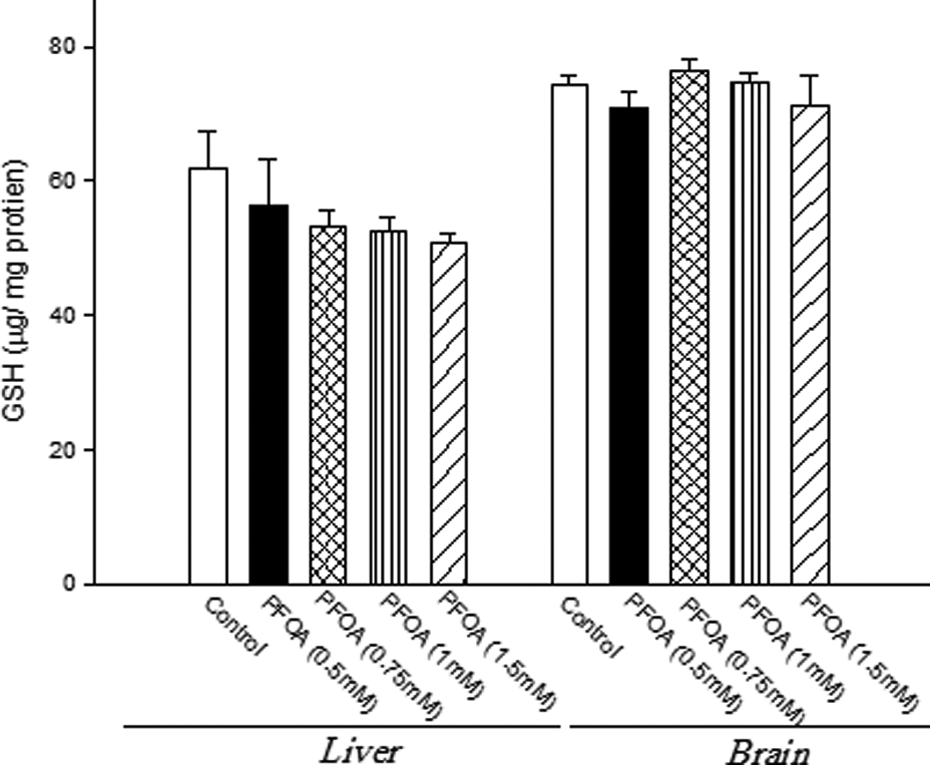
Effect of PFOA on mitochondrial GSH content. Liver and brain mitochondria (0.5 mg/ml) were incubated at 30°C for 1 h with PFOA (0.5, 0.75, 1, 1.5 mM). Values are presented as mean ± SEM (n = 3). PFOA: perfluorooctanoic acid; GSH: reduced glutathione.
Effect of PFOA on MMP
PFOA caused significant collapse of MMP in liver (0.5–1.5 mM) and brain (0.5–1.5 mM) samples (p < 0.05, Table 4). Based on our results the order of sensitivity of liver against PFOA-induced MMP collapse is higher than brain tissue.
The effect of PFOA on the MMP collapse (ΔΨ%) in isolated mitochondria.a

PFOA: perfluorooctanoic acid; MMP: mitochondrial membrane potential; CsA: cyclosporin A; BHT: butylated hydroxytoluene; Rh123: Rhodamine 123.
aMMP collapse (ΔΨ%) was measured by Rh123 as described in Materials and methods. The effect of PFOA (0.5, 0.75, 1 and 1.5 mM) on the MMP decrease in liver mitochondria was evaluated. Values are represented as mean ± SEM (n = 3).
bCalculated based on formula:
c p < 0.001: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA).
d p < 0.01: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA).
Effect of PFOA on mitochondrial ATP level
Treatment of mitochondria with all the concentrations of PFOA (0.5–1.5 mM) decreased ATP content in a concentration-dependant manner in both liver and brain samples (p < 0.05, Figure 5). Our results suggested that the decrease of ATP level on isolated liver mitochondria is higher than brain mitochondria, which indicates the mitochondrial dysfunction.

Effect of PFOA on ATP level in isolated liver and brain mitochondria. Liver and brain mitochondria (0.5 mg/ml) were incubated at 30°C for 1 h with various concentrations of PFOA (0.5, 0.75, 1, 1.5 mM). ATP level was evaluated as described in Materials and methods. Values represented as mean ± SEM (n = 3). ***p < 0.001: compared with control mitochondria. PFOA: perfluorooctanoic acid; ATP: adenosine triphosphate.
Effect of PFOA on mitochondrial swelling
Treatment of liver mitochondria with PFOA (0.5–1.5 mM) caused significant swelling of mitochondria in a time- and concentration-dependant manner (p < 0.05, Table 5). Likewise, swelling of brain mitochondria was significant in the presence of 0.75–1.5 mM concentrations of PFOA (p < 0.05, Table 5). Our results in mitochondrial swelling on isolated rat liver and brain mitochondria are in similar line with PFOA induced MMP collapse.
Effect of PFOA on the mitochondrial swelling in rat mitochondria.a

PFOA: perfluorooctanoic acid; CsA: cyclosporin A; BHT: butylated hydroxytoluene.
aMitochondrial swelling was measured by determination of absorbance at 540 nm as described in Materials and methods. Values represented as mean ± SEM (n = 3).
bCalculated based on the formula:
c p < 0.05: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA).
d p < 0.01: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA).
e p < 0.001: compared with control mitochondria (mitochondrial suspension treated with equivolume of buffer instead of PFOA).
Effect of PFOA on the release of cytochrome c
Treatment of liver mitochondria with 0.5 mM PFOA resulted in the release of cytochrome c from mitochondria as much as nearly 58% of the total mitochondrial cytochrome c. The effect was found to be concentration dependent (p < 0.05, Figure 6). In brain mitochondria, PFOA (0.75–1.5 mM) led to a significant expulsion of cytochrome c, compared to control group (p < 0.05, Figure 6). ATP depletion due to conversion of mitochondrial ATP synthase to ATPase, followed by substantial swelling and finally cytochrome c release. Our results confirmed that severity of cytochrome c release in liver is higher than in brain mitochondria.

Effect of PFOA on cytochrome c release. The amount of released cytochrome c from mitochondria was determined using rat/mouse cytochrome c ELISA kit as described in Materials and methods. Values (ng/mg protein) are represented as mean ± SEM (n = 3). ***p < 0.001: compared with control liver mitochondria; θθθ p < 0.001: compared with control brain mitochondria; PFOA: perfluorooctanoic acid; ELISA: enzyme-linked immunosorbent assay.
Discussion
Mitochondria, the major source of energy production in the cell, are the target organelle for toxicity mechanisms of xenobiotics such as heavy metals, chemicals and pesticides. The significance of using isolated mitochondria to evaluate the safety of chemicals has been fully recognized using liver tissue as well as those of heart, brain tissues. 11,25 Besides, mitochondrial damage could lead to oxidative stress and activation of apoptotic pathways, which ultimately has a fatal consequence to the cell. 26 It is supposed that assessment of mitochondrial function would be a reasonable practice which might help to decipher the mechanism behind the toxicity of a drug or chemical substance. 26 The role of mitochondria in the evaluation of toxic effects of xenobiotics is increasingly implicated and mitochondrial models are now widespread to determine safety and toxicity mechanisms of chemicals. 27 Besides, there is accumulating evidence supporting a direct relationship between mitochondria, oxidative stress and cell death. 28 Therefore, in this study, we have reported the assessment of the effects of PFOA on mitochondria isolated from rat liver and brain tissue with multi-parametric assays as new models for determination of safety and toxicity mechanisms of chemicals. 11
The concentration range of PFOA (0.5–1.5 mM) was selected on the basis of a series of pilot studies in our laboratory. The concentrations of PFOA used in this study might seem relatively high, regarding this fact that the accumulation possibility of PFOA in occupationally exposed workers cannot be neglected. Currently, environmental exposures are estimated to be on the tens of parts per billion concentration, and it is far lower concentrations of PFOA that produce any significant effects on mitochondria. But designation of an experimental model requires the number of animals to be smaller compared to the size of human populations at risk. Obtaining statistically valid results and determination of toxicity mechanisms from animals (in small groups) requires the administration of relatively large doses so that the effect will occur frequently enough to be detected. 29 Based on the recent results, we found significant elevation of ROS from 0.75 mM to 1.5 mM in liver mitochondria following exposure to PFOA similar to other studies. 11,30
Besides, complexes I and III of mitochondrial respiratory chain are the main sources of ROS production 31 and impairment in any component of the respiratory chain could cause mitochondrial dysfunction followed by oxidative stress and cell injury. 32 Based on these considerations, it was decided to figure out the precise site of ROS formation in PFOA-treated mitochondrial suspension. Our results showed the impaired activity of complexes I and III in liver mitochondria following incubation with PFOA (Table 3). In addition, it was also found that PFOA significantly inhibited mitochondrial succinate dehydrogenase (complex II) activity in a concentration-dependent manner with higher vigorous liver tissue (p < 0.05, Figure 2).
As an intracellular antioxidant defense factor, GSH protects cell from damaging effects of ROS such as hydrogen peroxide. It also maintains the thiol-containing proteins in their active form. GSH, in a non-enzymatic reaction with electrophiles, is oxidized to glutathione disulfide, but would not be released from the mitochondria and will be normally reduced back to GSH during the recovery of mitochondria from oxidative stress. 33 Therefore, it suggested that in isolated mitochondria, maintenance of the sulfhydryl groups in the reduced state only helps the closing of MPT pore and induction of oxidative stress. Based on our results, PFOA did not affect glutathione content in liver mitochondria (Figure 4). On the other hand, exposure of mitochondria to PFOA did not lead to any significant increase in MDA content as the marker of LPO (Figure 3). This study suggested that isolated brain mitochondria significantly increased LPO in brain mitochondria in a concentration-dependent manner. It seems that the higher concentration of polyunsaturated fatty acids in brain tissue is the main factor for the susceptibility of brain tissue to LPO. Mitochondrial lipids with neighborhood with early target of oxygen-free radicals caused higher production of MDA in brain tissue.
One of the key steps towards the mitochondria-mediated cell death is the opening of mitochondrial MPT pores. 34 Therefore, membrane depolarization is the primary consequence of MPT pore opening in in vitro or intact cells model based on the fluorescence changes of probes that are accumulated by mitochondria in response to the MMP collapse (Δψ). 35 Incubation of mitochondria with an excess of calcium or some known chemicals that induce oxidative stress leads to opening the MPT pores, which in turn 36 results in the passage of low-molecular-weight solutes across the inner membrane of mitochondria causing mitochondrial swelling, collapse of MMP and release of cytochrome c. Based on our results, exposure to PFOA of all the concentrations caused a significant MMP collapse in a time and concentration dependent manner (p < 0.05, Table 4). In addition, treatment of liver mitochondria with PFOA (0.5–1.5 mM) caused a significant swelling of mitochondria in a time and concentration dependant manner, which was higher in the liver (p < 0.05, Table 5).
Since mitochondrial swelling occurs subsequent to MMP collapse, our findings on mitochondrial swelling confirms the role of MPT pore opening in PFOA-induced mitochondrial dysfunction. Furthermore, we observed that cyclosporine A (CsA, 1 μM) as an inhibitor of MPT pore and butylated hydroxytoluene (BHT, 20 μM) as an antioxidant largely inhibited MMP collapse. Our findings are in agreement with those of other studies regarding the toxicity of fluorochemicals. Panaretakis et al. in their study on HepG2 cells found a dissipation of MMP following incubation with PFOA, which was reversed by pretreatment with CsA. 12 Starkov and Wallace also have reported some similar findings on mitochondria isolated from rat liver. They found that treatment of mitochondria with some perfluorinated derivatives induced MMP collapse and mitochondrial swelling. 37
ATP is mainly produced through the oxidative phosphorylation in mitochondrial respiratory chain and impairment in electron transport chain leads to decline in ATP synthesis. In addition, it can be deducted that exposure to PFOA leads towards mitochondrial MMP collapse. Based on the existing reports, in conditions of declined MMP, the F1F0-ATP synthase, in order to maintain MMP can reverse its catalytic activity and hydrolyze ATP. This leads to a further reduction of mitochondrial ATP content. 38 Therefore, we examined if PFOA could have any effect on mitochondrial ATP level. As shown in Figure 5, treatment of mitochondria with all the concentrations of PFOA significantly decreased ATP content (p < 0.05). Therefore, the inhibitory effect of PFOA on respiratory chain is reconfirmed by the decreased levels of ATP following incubation with PFOA. Besides, it suggested that decrease of ATP level on isolated liver mitochondria is higher than that on brain mitochondria. It supposed that any damage that impairs the function of the mitochondrial respiratory chain system might also have an impact on cell viability. Therefore, mitochondria are the important defence barrier or protection system in cells for detoxifying and repairing ROS-induced damage. 28 Our results showed a considerable release of cytochrome c from liver mitochondria. It is well known that release of cytochrome c to cytosol starts a complex of further events known as cell death signalling (Figure 6). The results of our experiments on brain mitochondria showed both similarities and differences in comparison with those obtained on liver mitochondria. Assessment of ROS generation indicated that exposure of brain mitochondria to PFOA led to significant ROS formation (Tables 1 and 2). Our investigations in brain mitochondria showed that PFOA interfered with the activity of complexes I and III, similar to what we observed in liver mitochondria (Table 3). However, succinate dehydrogenase (complex II) activity on isolated brain mitochondria was affected only by the highest concentration of PFOA (1.5 mM, Figure 2). It is concluded that sensitivity of liver against PFOA toxicity is higher than brain tissue. It suggested that the main site of MTT reduction is mitochondrial complex II activity and succinate dehydrogenase is responsible for most cellular reduction and is the criteria for evaluation of electron transfer chain defect.
Similar to liver mitochondria, exposure to PFOA did not alter GSH content (Figure 4). However, PFOA (0.5–1.5 mM) significantly increased (LPO) in brain mitochondria (Figure 3). The observed difference between the two organs could be explained in light of the fact that brain mitochondria contain a high level of unsaturated fatty acids such as cardiolipin that are susceptible to LPO. Previous reports have suggested that there is a close relationship between the concentration of polyunsaturated fatty acids and susceptibility to LPO. 39
Following our assessment of MMP and monitoring of brain mitochondrial swelling, similar results to those obtained from liver mitochondria were found. Incubation of brain mitochondria with PFOA significantly decreased MMP and increased mitochondrial swelling (Tables 4 and 5). Pretreatment of mitochondria with CsA and BHT inhibited MMP collapse, which suggests the role of MPT induction in the observed decline in MMP following incubation with PFOA.
The predominant physiological function of mitochondria is the generation of ATP by oxidative phosphorylation. Evaluation of ATP level showed significant decrease in ATP content following incubation of brain mitochondria with PFOA (Figure 5). The decreased ATP level could be a consequence of PFOA-induced impairment of mitochondrial respiratory chain and MMP collapse as described earlier. It has been proved that ATP level is a determinant factor for the mode of cell death. This is because apoptosis is a complex biochemical process requiring ATP. Therefore, in conditions with a high depletion of ATP source, the cell is shifted towards necrotic cell death pathway. 40 In the mitochondrial samples isolated from rat liver, the 0.5, 0.75, 1 and 1.5 mM concentrations of PFOA significantly inhibited the production of ATP up to 53, 63, 67, 83% (p < 0.001), respectively. Similarly, in the brain mitochondria, there was a significant decrease in ATP level (31–42%) following the exposure with different concentrations of PFOA. Comparison of the results in liver and brain mitochondria showed the higher depletion of ATP level in isolated liver mitochondria (53–83% reduction in ATP level). It could therefore be proposed that PFOA in low concentrations in liver and in all the concentrations in brain would induce apoptosis, whilst necrosis would be quite likely in higher concentrations due to severe depletion of ATP level in 0.75–1.5 mM in liver mitochondria.
Finally, the results of cytochrome c assays revealed that incubation of brain mitochondria with PFOA led to a significant expulsion of cytochrome c (Figure 6). Cytochrome c, with association of cardiolipin, is attached to the inner membrane of mitochondria where it can reversibly interact with complexes III and IV of the respiratory chain. It has also been established that decreased content of cardiolipin (which could be caused by LPO) could lead to the detachment of cytochrome c from inner mitochondrial membrane and facilitate the release of cytochrome c to cytosol. 41 Therefore, LPO could ultimately result in the induction of cytochrome c release from isolated rat brain mitochondria upon exposure with PFOA. Besides, it is suggested that in mitochondrial level CsA could be blocked by the release of cytochrome c from mitochondria by non-MPT pathway via inhibition of structural remodelling of cristae on inner mitochondrial membrane. 42
Conclusion
Mitochondrial dysfunction is a pathogenic mechanism that leads to toxicities in organisms especially in the liver and brain. The results of this study indicate that PFOA has been affective on some vital biochemical functions of mitochondria. In liver mitochondria, PFOA increased ROS generation, interfering with the activities of complexes I, II and III in mitochondrial respiratory chain; caused MMP collapse and mitochondrial swelling; decreased ATP level; and caused the release of cytochrome c from mitochondria. The latter event has been established to trigger apoptotic pathways in the cell. In brain mitochondria, PFOA had fairly similar effects on ROS generation, activity of mitochondrial complexes I and III, MMP, mitochondrial swelling, ATP level and release of cytochrome c. PFOA did not have a significant effect on GSH content in both liver and brain mitochondria. However, incubation of brain mitochondria with PFOA increased LPO, whilst the activity of mitochondrial complex II remained intact. PFOA caused the release of cytochrome c in both liver and brain mitochondria which could ultimately lead to cell death. As a general trend, we observed that PFOA affected the functions of liver mitochondria more than that of brain mitochondria. Therefore, it could be suggested that liver mitochondria are more susceptible to the toxic effects of PFOA. Due to the fact that PFOA has toxic effects on the mitochondria isolated from liver and brain tissue, this study suggests that mitochondrial toxicity could be a plausible mechanism for the toxic effects of this fluorochemical compound on liver and brain function.
Footnotes
Conflict of interest
The authors declared no conflicts of interest.
Funding
This work was supported by the Deputy of Research of Zabol University of Medical Sciences (Grant No. 119-92).