
Abstract
Amyloid depositions of proteins play crucial roles in a wide variety of degenerative disorders called amyloidosis. Although the exact mechanisms involved in amyloid-mediated cytotoxicity remain unknown, increased formation of reactive oxygen species (ROS) and nitrogen species and overproduction of pro-inflammatory cytokines are believed to play key roles in the process. In that regard, we investigated the effect of apigenin, a common dietary flavonoid with high antioxidant and anti-inflammatory properties on potential factors involved in cytotoxicity of human insulin amyloids. Pretreatment of SK-N-MC neuroblastoma cells with apigenin increased cell viability and reduced the apoptosis induced by insulin fibrils. In addition, apigenin attenuated insulin fibril-induced ROS production and lipid peroxidation. Our result also demonstrated that pretreatment of the fibril-affected cells with apigenin caused an increase in catalase activity and the intracellular glutathione content along with reduction in nitric oxide production and nuclear factor κB, tumor necrosis factor α, and interleukin 6 gene expression based on real-time polymerase chain reaction evaluation. In accordance with these results, apigenin could be a promising candidate in the design of natural-based drugs for treatment or prevention of amyloid-related disorders.
Keywords
Introduction
Aggregation of proteins, mainly in the form of amyloid-like fibrils, is well accepted as a key pathogenic feature of more than 20 different clinical syndromes, including Alzheimer’s, Parkinson’s, and Huntington’s diseases, type II diabetes, and prion diseases. 1 During these events, normally folded soluble proteins form misfolded intermediates and assemble into fibrillar deposits which lead to cell damage and organ dysfunction. The amyloid fibril formation is not restricted to disease-related proteins. Many other proteins also have the potential to form amyloid aggregates in vitro under partially denaturing conditions. 2 Although the structural homology and functional similarity of these amyloid-forming proteins and peptides vary widely, most amyloid fibrils appear to share common characteristics, such as the ability to bind Congo red and thioflavin-T, exhibiting cytotoxicity effects, and sharing a common cross-β structure. 3
Although the precise mechanisms involved in amyloidogenic assemblies-mediated cytotoxicity remain unknown, destabilization of cellular membrane, increased formation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and overproduction of pro-inflammatory cytokines are believed to play key roles in the process. 4,5 It has been demonstrated that amyloids can bind to the cell membrane to form channels or pores that disrupt ion homeostasis, hence leading to cellular dysfunction. 5 On the other hand, nitrostative and oxidative damage has been reported in different amyloidosis diseases such as Alzheimer and Parkinson. 6,7 Numerous studies have provided evidence that amyloids are involved in ROS generation. Overproduction of ROS, under oxidative stress conditions, is known to lead to membrane lipid peroxidation, inflammatory responses, and induction of proapoptotic signals such as caspase 3. 8,9 Moreover, amyloid fibrils can induce nitric oxide (NO) generation by overexpression or overactivation of NO synthase (NOS). 5,10 Although the mechanism of NO-mediated cytotoxicity remains uncertain, studies have shown that NO can modify protein function by nitrosylation and nitrotyrosination, inhibit mitochondrial respiratory complexes, participate in organelle fragmentation, and mobilize zinc from internal stores. 11,12 Another potential mechanism of cytotoxicity of amyloid fibrils may involve inflammatory responses. Inflammation is considered both as a consequence and as a cause of oxidative stress. It has been demonstrated that ROS and RNS act as signals to activate pro-inflammatory genes such as tumor necrosis factor α (TNF-α) and nuclear factor κB (NF-κB) and thereby causing chronic inflammation. In turn, chronic inflammation leads to generation of ROS and RNS. 13 Regarding this fact, the inhibition of amyloid formation, disruption of the formed amyloids, and reducing amyloid-induced oxidative and nitrostative stress and pro-inflammatory cytokines production are potential therapeutic and preventive strategies for amyloid-related degenerative diseases.
Apigenin (4′,5,7-trihydroxyflavone; Figure 1), a member of the flavone family, is found in many fruits, vegetables, and nuts. 14 Apigenin has been in the center of focus in recent years as a health-promoting agent compared to other structurally related flavonoids. 15 A number of biological effects of apigenin in numerous systems in vitro as well as in vivo are related to its potent antioxidant capacity and its role in scavenging free radicals. 15 Moreover, it possesses various pharmacological effects such as antimutagenic, antiviral, antitumor, and anti-inflammatory activities. 16–18 Apigenin inhibits the production of pro-inflammatory mediators such as TNF-α and interleukin 6 (IL-6) in several cell lines through NF-κB signalling pathway. 19,20 In addition, we have recently reported that apigenin exhibited an inhibitory effect on the amyloid formation of human insulin. 21

Chemical structure of apigenin.
Regarding the proven roles of ROS in the production of pro-inflammatory agents and the role of protein aggregates in ROS production and the antioxidant activity of apigenin, we hypothesized that attenuation of ROS level by apigenin would lead to lower levels of pro-inflammatory factors and thus lower cytotoxicity on neuroblastoma cells exposed to insulin fibrils.
Materials and methods
Materials
The cell culture medium (Rosewell Park Memorial Institute (RPMI) 1640), penicillin–streptomycin, and fetal bovine serum (FBS) were purchased from Gibco BRL (Life technology, Paisley, Scotland). Apigenin, 3-(4,5-dimethyl tiazol-2-yl)-2,5 diphenyl tetrazolium bromide (MTT), Hoechst 33258, and 5,5′-dithiobisnitro benzoic acid (DTNB) were obtained from Sigma–Aldrich (St Louis, Missouri, USA). 2′,7′-Dichlorofluorescin diacetate (DCFH-DA) was purchased from Invitrogen (Carlsbard, California, USA). Human insulin was a generous gift from Dr Byrn (Purdue University). Thiobarbituric acid (TBA), hydrogen peroxide (H2O2), and dimethyl sulfoxide (DMSO) were purchased from Merck (Darmstadt, Germany). All primers were obtained from Bioneer (Chungwon, Korea). All other chemicals used were of analytical grade.
Preparation of insulin fibrils
Human insulin sample solutions (4 mg ml−1) were prepared in glycine–hydrochloric acid buffer (50 mM, pH 2) containing 0.02% sodium azide. The concentration of insulin was determined using an extinction coefficient of 1.0 for 1 mg ml−1 at 276 nm. 22 To induce the production of the amyloid structure, insulin solutions were incubated for 12 days at 50°C in a water bath without agitation. 23 Preparation of fibrillar insulin was verified using transmission electron microscopy (TEM; data not shown). The fibril concentration was determined by subtracting the insulin monomer concentration, measured in the supernatant following centrifugation at 15,000 r min−1 for 60 min, from the original solution of insulin monomer concentration.
Cell culture
Human SK-N-MC neuroblastoma cells were obtained from Pasteur Institute of Iran (Tehran, Iran). SK-N-MC cells were cultured at a density of 25 × 103 cells ml−1 in RPMI 1640 medium supplemented with 10% FBS, penicillin (100 U ml−1), and streptomycin (100 μg ml−1) and incubated under humidified atmosphere of 5% carbon dioxide at 37°C. Apigenin was dissolved in a minimum amount of DMSO and then diluted with the culture medium to get the desired concentration. The concentration of DMSO in the culture medium has been kept lower than 0.1%, and the control cells have been treated with the vehicle containing the same amount of DMSO.
Measurement of cell viability
To evaluate the protective effect of apigenin against insulin fibril toxicity, SK-N-MC cells were seeded in a 96-well plate, and after 24 h, they were pretreated with various concentrations of apigenin (5–20 μM) for 3 h followed by exposure to insulin fibrils (10 μM) for an additional 24 h. After incubation, the viabilities of SK-N-MC cells were determined using the MTT assay. Briefly, the cells were treated with 10 μl MTT (5 mg ml−1) for 4 h. Then, the supernatant was removed and the formazone crystals were solubilized with 200 μl DMSO, and the absorbance at 570 nm was read with an enzyme-linked immunosorbent assay (ELISA) reader (Expert 96, Asys Hitch, EC Austria). Results were expressed in percentage of survival relative to the control cell samples.
Fluorescence microscopy evaluation of the apoptotic cells
The SK-N-MC cells were seeded in 12-well plates. They were preincubated with apigenin (10 and 20 μM) for 3 h and then were treated with insulin fibrils (10 μM) for a time course of 24 h. Apoptotic cells were characterized morphologically by staining the cells with Hoechst 33258 followed by fluorescence microscopy inspection. After treatment, the cells of each well were washed twice with phosphate-buffered saline (PBS) and adjusted to a cell density of 1 × 106 cells ml−1 of PBS. Hoechst 33258 solution (1 mg 1 ml−1 double-distilled water) was added to the cell suspension in a final concentration of 100 μg ml−1. The cellular morphology was evaluated using an Axoscope 2 plus fluorescence microscope from Zeiss (Germany). All experiments were repeated three times, and the number of stained cells was manually counted in 10 randomly selected fields. The extent of apoptosis was then expressed as a percentage of the total cell count.
Assessment of intracellular ROS
The intracellular ROS level was determined using DCFH-DA dye. 24 This dye is a stable compound that readily diffuses in to the cells, and it is then hydrolyzed by the intracellular esterase to yield DCFH which is trapped within the cells. The intracellular ROS oxidize DCFH to a highly fluorescence compound 2′,7′-dichlorofluresin. Thus, the fluorescence intensity is proportional to the amount of intracellular ROS produced in the cells. Treated and untreated cells were incubated with 10 μM DCFH-DA at 37°C for 60 min and then washed twice with PBS. Then, ROS was quantitated using a flow cytometer (PartecPAS, Germany).
Determination of lipid peroxidation
The extent of lipid peroxidation was measured by the TBA reactive substances (TBARS) assay. 25 The method is based on the spectrophotometric measurement of the pink color generated by the reaction of TBA with TBARS mainly malondialdehyde (MDA). The method involved heating the cell lysate with TBA reagent for 20 min in a boiling water bath. TBA reagent contains 20% trichloroacetic acid, 0.67% TBA, and 25% acetic acid. After cooling, the solution was centrifuged at 3000 r min−1 for 10 min, and the precipitate obtained was removed. The absorbance of the supernatant was determined at 532 nm with respect to the blank solution. The protein concentration was determined by Lowry’s method. 26 The concentration of MDA was calculated based on the absorbance coefficient of the TBA-MDA complex (ε = 1.56 × 105 cm−1 M−1) and expressed as nanomoles per milligram protein.
Catalase activity assay
The catalase activity was measured by the method described by Aebi. 27 Briefly, the rate of decrease in H2O2 was measured spectrophotometrically at 240 nm in a reaction mixture containing 10 mM H2O2, 50 mM monopotassium phosphate and 50 μM of each cell lysate. Activity of catalase was calculated as ×10−1 k mg−1 protein, where k represents the rate constant of the first-order reaction of catalase.
Measurement of intracellular GSH
The concentration of glutathione (GSH) was determined in the whole cell lysate using DTNB method at 412 nm. 28 Briefly, 0.5 ml of each cell lysate was precipitated with 1 ml of sulfosalicylic acid (4% w/v). The precipitate was removed by centrifugation. Each supernatant (0.5 ml) was mixed with 0.1 ml DTNB (4 mg ml−1) and 0.9 ml phosphate buffer (0.1 M, pH 7.4). The yellow color developed was read at 412 nm. Reduced GSH was expressed in micrograms per milligram protein.
Determination of NO production
NO level can be determined spectrophotometrically by measuring the accumulation of its stable degradation products, nitrite and nitrate. The nitrite accumulation in culture medium was determined using the Griess reagent (1% sulfanilamide in 5% phosphoric acid and 0.1% 1-naphtylethylenediamine). 29 To this, 100 μl of culture medium was mixed and incubated with 100 μl of Griess reagent for 15 min at room temperature, and the absorbance was measured at 540 nm using an ELISA reader (Expert 96, Asys Hitch, EC Austria). Serial dilutions of sodium nitrite were used as the standards.
Determination of mRNA level using real-time PCR
Total RNA was isolated from cultured cells using the TRIzol reagent according to the manufacturer’s protocol (Invitrogen, Carlsbard, California, USA). The RNA concentration and the extent of its purity were determined spectrophotometrically at 260 nm and by the A
260/A
280 ratio, respectively. Then, 1 μg of RNA from each sample was subjected to reverse transcription using the Prime Script RT reagent kit (Takara Bio Inc., Otsu, Japan). Real-time polymerase chain reaction (PCR) was performed with a light cycler instrument (Roche Diagnostics GmbH, Mannheim, Germany) using SYBR Premix Ex Taq technology (Takara Bio Inc., Otsu, Japan). A SYBR Green master mix (10 μl) was added to 2 μl of complementary DNA samples, 0.5 μl of forward and reverse primers (10 pmol), and 7 μl of nuclease-free water to conduct PCR in 20 μl of reaction mixture. Thermal cycling conditions involved an initial activation step for 30 s at 95°C followed by 45 cycles including a denaturation step for 5 s at 95°C and a combined annealing/extension step for 30 s at 60°C. Melting curves were analyzed to validate single PCR product of each primer. Hypoxanthine phosphoribosyltransferase 1 (HPRT1) was used as a normalize, and the fold change in expression of each target messenger RNA (mRNA) relative to HPRT1 was calculated based on
Sequences of primers used for real-time PCR.

TNF-α: tumor necrosis factor α; IL-6: interleukin 6; IL-1β: interleukin 1β; NF-κB: nuclear factor κB; HPRT: hypoxanthine phosphoribosyltransferase.
Statistical analyses
All data are presented as means ± SD. The mean values were calculated based on the data taken from at least three independent experiments using freshly prepared reagents. The statistical significances were achieved when p < 0.05.
Results
Apigenin protects SK-N-MC cells against insulin fibril-induced cytotoxicity
The effect of apigenin on human insulin fibril cytotoxicity was investigated using MTT assay. As shown in Figure 2(a), 10 μM amyloid fibrils decreased the viability of SK-N-MC cells to about 66% of control. However, apigenin protected the cells against the cellular damage induced by insulin amyloids in a dose-dependent manner. The survival level was increased to 69%, 73%, 84%, and 93% when the cells were pretreated with 2, 5, 10, and 20 μM apigenin for 3 h, respectively (Figure 2(a)).

Effect of apigenin on insulin fibril-induced cytotoxicity (a) and apoptosis (b, c). SK-N-MC cells were treated with different concentrations of apigenin for 3 h, followed by exposure to insulin fibrils (10 μM) for 24 h. The extent of apoptosis was calculated as described in the Fluorescence Microscopy Evaluation of the Apoptotic Cells section. Data represent means ± SD (n = 3). *p < 0.05: significantly different from untreated control cells. **p < 0.05: significantly different from insulin fibril-treated cells.
Apigenin prevents insulin fibril-induced apoptotic cell death
To further study the cell protecting effect of apigenin, Hoechst 33258 staining technique was used to evaluate the occurrence of apoptosis. As shown in Figure 2(c), viable cells are uniformly blue, whereas apoptotic cells are blue and contain bright blue dots in their nuclei as a consequence of chromatin condensation and nuclear fragmentation. The extent of apoptotic cell death for untreated and cells treated with apigenin (20 μM) for 24 h were lower than 5%. The number of apoptotic cells increased to 21.8% upon exposure to insulin fibrils (10 μM) for 24 h. However, pretreatment with apigenin decreased the extent of apoptotic cells relative to insulin fibril-treated cells, dose dependently (Figure 2(b)).
Apigenin attenuates insulin fibril-induced increase in intracellular ROS generation
In order to examine whether the anti-apoptotic effect of apigenin was related to the alteration of intracellular redox status of SK-N-MC cells, the intracellular ROS level was measured using DCFH-DA staining approach. According to the flow cytometry analyses, the generation of intracellular ROS in SK-N-MC cells increased to 31.05% after treatment with 10 μM insulin fibrils compared with the ROS level of the control cells (5.47%). However, as it is evident from Figure 3, pretreatment with 10 and 20 μM of apigenin reduced ROS generation to 20.55% and 10.51%, respectively.
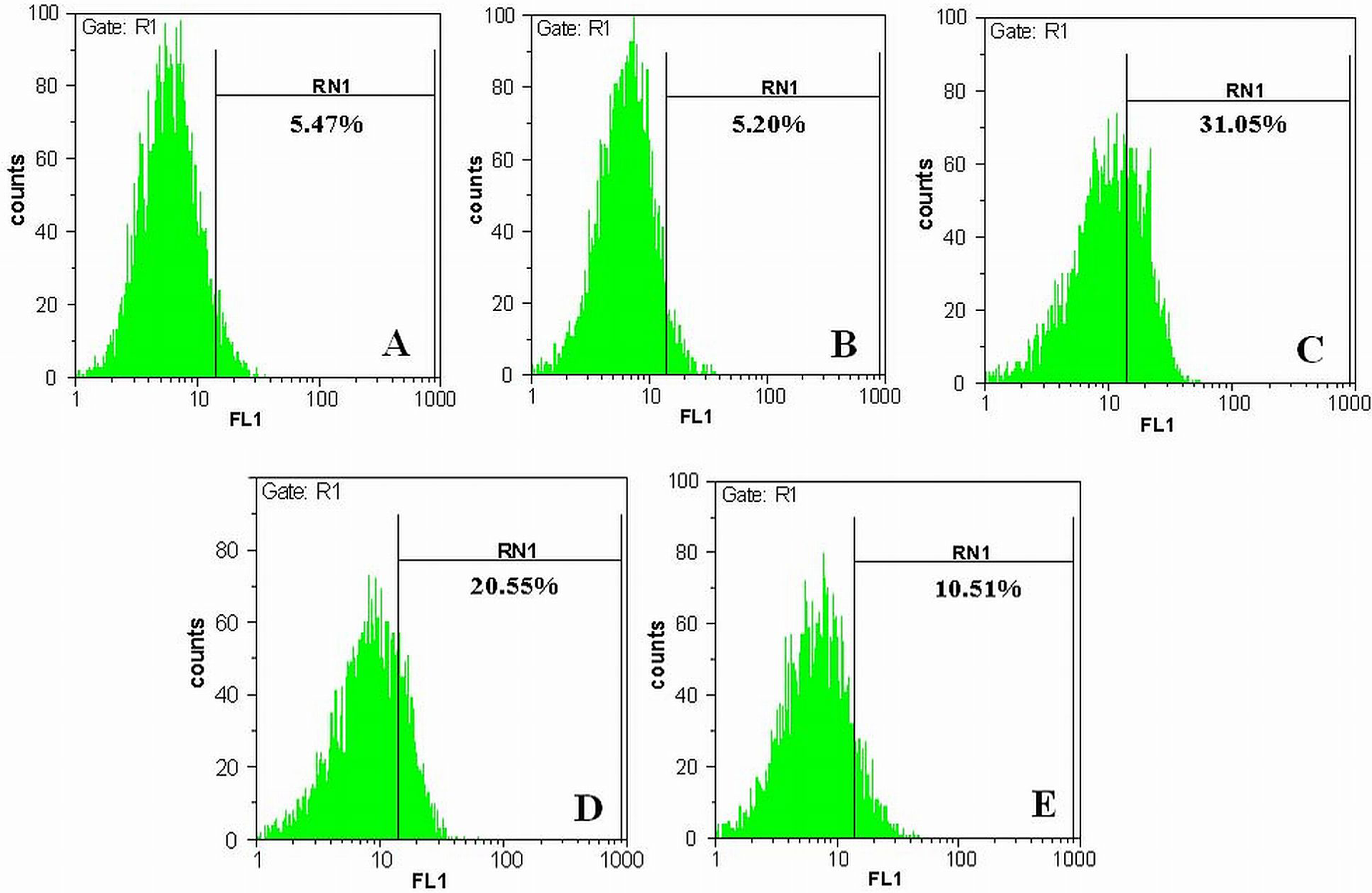
Inhibitory effects of apigenin on intracellular ROS production induced by insulin fibrils. The production of ROS was monitored by DCFH-DA staining, and the fluorescence intensity of 10,000 cells was analyzed using flow cytometry technique. Untreated control cells (a), cells treated with 20 μM apigenin (b), cells treated with 10 μM insulin fibrils (c), cells treated with 10 and 20 μM apigenin for 3 h, followed by exposure to 10 μM insulin fibrils (d and e). ROS: reactive oxygen species; DCFH-DA: 2′,7′-dichloroflourescin diacetate.
Apigenin represses insulin fibril-induced lipid peroxidation
To further confirm the free radical scavenging activity of apigenin among the insulin fibril-treated cells, TBARS levels were measured. Among these reactive substances, MDA is a unique end product of lipid peroxidation that usually represents the extent of lipid peroxidation. After 1 day of incubation with 10 μM insulin fibrils, the MDA level was greatly increased compared with the control cells (0.89 nmol mg−1 protein in control cells vs. 2.77 nmol mg−1 protein in fibril-treated cells). This increase was suppressed by the addition of 10 and 20 μM of apigenin. As shown in Figure 4(a), the extent of MDA formation was reduced to 2.02 and 1.12 nmol mg−1 protein when the cells were pretreated with 10 and 20 μM apigenin, respectively.

Effects of apigenin on lipid peroxidation, GSH level, catalase activity and NO production in insulin fibril-treated cells. SK-N-MC cells were treated with 10 and 20 μM apigenin for 3 h, followed by exposure to insulin fibrils (10 μM) for 24 h. Data represent means ± SD (n = 3). *p < 0.05: significantly different from untreated control cells . **p < 0.05: significantly different from insulin fibril-treated cells. GSH: glutathione; NO: nitric oxide.
Apigenin increases intracellular GSH level in the insulin fibril-treated cells
GSH has an essential role in scavenging free radicals and thus it plays a key role in maintenance of intracellular oxidative balance. The intracellular GSH content of SK-N-MC cells was reduced by 28.3% relative to untreated control following exposure of the cells to 10 μM insulin fibrils for 24 h. The GSH contents of SK-N-MC cells, pretreated with 10 and 20 μM apigenin, increased to 76.4% and 88.0%, respectively, relative to the fibril-treated cells (Figure 4(b)).
Apigenin increases catalase activity in the insulin fibril-treated cells
To further elucidate the mechanism underlying the antioxidant function of apigenin, the activity of catalase, as one of the most responsive antioxidant enzymes of the biological system, was determined among the insulin fibril-treated cells. As shown in Figure 4(c), the activity of catalase decreased by 54.8% among cells treated with insulin fibrils (10 μM). However, pretreatment of the cells with apigenin at concentrations of 10 and 20 μM increased the catalase activity by 20.7% and 39.1%, respectively, relative to cells treated solely with insulin fibrils.
Apigenin reduces insulin fibril-induced NO production
Nitrite accumulation in culture medium was measured using the Griess method as an indirect measurement of RNS production. As shown in Figure 4(d), exposure to 10 μM insulin fibrils increased the NO production by 42.7%, relative to untreated control cells. Pretreatment of the cells with apigenin at concentrations of 10 and 20 μM decreased the NO production by 16.1% and 24.2%, respectively, relative to fibril-treated cells.
Apigenin reduces insulin fibril-induced NF-κB and pro-inflammatory cytokines expression
To investigate the effect of apigenin on the pro-inflammatory cytokines in insulin fibril-treated cells, TNF-α, IL-1β, and IL-6 gene expressions were measured by real-time PCR analysis. As shown in Figure 5, in fibril-treated cells, the TNF-α, IL-1β, and IL-6 mRNA levels have increased by 1.3-, 1.6-, and 1.8-fold, respectively, compared with the untreated control cells. However, pretreatment with 20 μM apigenin decreased the TNF-α, IL-1β, and IL-6 gene expressions by 26%, 8%, and 44%, respectively, relative to cells treated solely with insulin fibrils. In addition, our data showed that treatment of SK-N-MC cells with 10 μM insulin fibrils induced expression of NF-κB (1.47-fold compared with untreated control cells). As shown in Figure 6, pretreatment of the cells with apigenin at concentration of 20 μM decreased the NF-κB mRNA level by 21% relative to fibril-treated cells.

Effect of apigenin on insulin fibril-induced pro-inflammatory cytokines expression. The relative mRNA expression of TNF-α, IL-1β, and IL-6 were measured using real-time PCR after normalizing the C ts of each sample against their corresponding HPRT. Data represent means ± SD (n = 3). *p < 0.05: significantly different from untreated control cells. **p < 0.05: significantly different from insulin fibril-treated cells. mRNA: messenger RNA; TNF-α: tumor necrosis factor α; IL-1β: interleukin-1 β; IL-6: interleukin-6; C t: cycle threshold; HPRT: hypoxanthine phosphoribosyltransferase.

Effect of apigenin on insulin fibril-induced NF-κB gene expression (a). Predicted cytoprotective mode of action of apigenin (b). The relative mRNA expression of NF-κB was measured using real-time PCR after normalizing the C ts of each sample against their corresponding HPRT. Data represent means ± SD (n = 3). *p < 0.05: significantly different from untreated control cells. **p < 0.05: significantly different from insulin fibril-treated cells. NF-κB: nuclear factor κB; mRNA: messenger RNA; C t: cycle threshold; HPRT: hypoxanthine phosphoribosyltransferase.
Discussion
It has been demonstrated that oxidative stress promote the production of a wide spectrum of pro-inflammatory cytokines including TNF-α and interleukins (IL-1β and IL-6) through NF-κB signalling pathway, which then induce cellular death. 13 TNF-α is a pleiotropic cytokine capable of inducing proliferation, inflammation through production of several inflammatory mediators, and cell death among many other effects. 33 IL-1β, as one of the best characterized cytokines, is expressed by a variety of cell types. This cytokine is involved in pathogenesis of a number of inflammatory-associated diseases such as diabetes, Alzheimer’s, and Parkinson’s diseases. 34 IL-1β can induce expression of a number of other inflammatory-associated proteins that form an amplified cascade of inflammatory response. In addition, it has been demonstrated that TNF-α and IL-1β could upregulate cyclo-oxygenase 2 (COX-2) and the inducible NOS (iNOS) expression, which would increase the production of free radicals such as NO and ROS. 33,35 The other important pro-inflammatory cytokine that can be induced under oxidative stress is IL-6 with a wide range of biological activities. 36 Since overproduction of pro-inflammatory cytokines, with subsequent ROS production has been observed in many inflammatory-associated diseases, it is anticipated that attenuation of inflammatory mediator levels could constitute the basis of a strategy for protection of cells against amyloid-induced oxidant damages.
In this study, human insulin was used as a model protein for production of amyloid fibrils. In vitro studies have shown that under denaturing conditions, such as acidic pH and high temperature, insulin molecules aggregate into amyloid fibrils. 37 In vivo, insulin amyloid deposits are believed to be the main player in the pathogenesis of the clinical conditions observed in diabetic patients, called insulin injection amyloidosis. 38 Additionally, amyloid fibrillation may cause problems during the production, storage, and delivery of insulin as a pharmaceutical compound. 37 The amyloid form of insulin becomes therapeutically ineffective, and the injection of such species probably causes unwanted/adverse immune responses. 39 Recently, serum samples from patients with Parkinson’s disease have been found to display an autoimmune response to insulin oligomers and fibrils, possibly indicating the presence of insulin aggregates in sera of the patients. 40 Moreover, the fibrillar species of insulin have been shown to be toxic to several cell types in vitro. 41,42
One promising strategy for reducing the cytotoxicity of amyloids could be either to attenuate the extent of protein aggregation, or to destabilize the already formed fibrils, to lower the surplus levels of ROS, or to lower the levels of relevant pro-inflammatory mediators. These could be done by using appropriately designed organic molecules, possessing appropriate hydrophobicity, free radical scavenging activity, and suppressive effects on the expression of inflammatory mediators. Our results indicated that apigenin, a natural flavonoid, is such a novel compound. We have recently reported that apigenin had an inhibitory effect on the amyloid formation of human insulin. 21 Hydrophobic interactions, aromatic stacking, and hydrogen binding are suggested to be the driving force in the anti-amyloidogenic effects of apigenin. 21 In this study, we evaluated the protective effects of apigenin on the cytotoxic role of the human insulin fibrillar structures. Our result demonstrated that treatment of the cells with the insulin fibrils (10 μM) for 24 h decreased the cell viability by 34% relative to untreated control cells. Parallel to the literature data (using other protein fibrils), our results indicated higher incidence of apoptosis (6.1-fold) among the cells treated with insulin fibrils, relative to control cells. 43 However, pretreatment with apigenin significantly increased cell viability and reduced the apoptosis induced by the insulin fibrils.
On the other hand, several studies have emphasized that oxidative stress governs the cytotoxicity of amyloid fibrils. 5–7 It has been shown that treatment with antioxidant compounds can provide protection against amyloid toxicity. 8,43,44 The fibrillar aggregates lead to oxidative stress through different routes including increase in ROS level, decrease in activity or expression of antioxidant enzymes, and dysfunctioning of mitochondria. The excess accumulation of ROS such as H2O2, hydroxyl radicals, and superoxide anions promotes oxidative damages to vital structural and functional molecules such as nucleic acids, proteins, and lipids leading to harmful effects on the biological system. 45,46 Lipid peroxidation is an important biological consequence of oxidative stress which is drastically increased in amyloid-treated cells. 44,47 In this study, apigenin prevented ROS production and lipid peroxidation induced by insulin amyloids. The antioxidant property of apigenin could be attributed to its free radical scavenging activity that has been demonstrated in many studies. 48–50 The presence of hydroxyl group at C-5 and C-7 and a double bond between C-2 and C-3 are required for ROS scavenging activity of apigenin. 51
Endogenous antioxidants have the capability to prevent the uncontrolled formation of free radicals and ROS and/or to inhibit their reaction with biological molecules. These antioxidants include mainly enzymes such as superoxide dismutase and catalase and nonenzymatic antioxidants such as GSH. It has been reported that the levels of these antioxidants decrease in the fibril-treated cells. 43 The decrease in GSH level is probably due to its increased utilization by the cells to counteract the increased level of free radicals. In our study, apigenin reversed the decreased intracellular GSH levels induced by insulin fibrils. This possibly indicates that apigenin can either increase the biosynthesis of GSH or reduce the extent of oxidative stress leading to less GSH degradation, or it may have both effects. On the other hand, the activity of the catalase was lower in the insulin fibril-treated cells relative to the control cells. The decreased activity of this enzyme can lead to surplus production of H2O2 in biological systems, which in turn will generate hydroxyl radicals involved in initiation and propagation of lipid peroxidation. 52 However, pretreatment of the fibril-affected cells with apigenin significantly increased the catalase activities. These results indicate that apigenin can indirectly decrease oxidative damages by activating endogenous defense system.
Moreover, numerous studies have provided evidences that amyloid aggregates are involved in NO generation. 7,53 NO exhibits neurotoxic properties and plays a pivotal role in the cascade of events leading to neuronal death. 11 Our data showed that the release of NO increased in insulin fibril-treated SK-N-MC neuroblastoma cells. This result may attribute to overexpression or overactivation of NOS. It has been demonstrated that amyloid aggregates can induce the expression of iNOS enzyme through the activation of NF-κB. 10 On the other hand, amyloid fibrils with overactivation of N-methyl-D-aspartate receptors cause excessive calcium ion influx and activation of neuronal NOS, resulting in excessive NO production. 5 However, in our study, pretreatment of the cells with apigenin decreased the NO production induced by insulin fibrils. This inhibitory effect of apigenin might be due to its strong antioxidant and anti-inflammatory activity. It has been demonstrated that oxidative stress and pro-inflammatory cytokines can enhance the transcription of iNOS gene via NF-κB activation. 35,54 In addition, Liang et al. have reported that apigenin is the most potent inhibitor of activity and expression of iNOS among the various flavnoids and thereby inhibits the production of NO in mouse macrophages. 55
Our results also showed that apigenin could decrease TNF-α and IL-6 gene expressions induced by insulin fibril, suggesting that apigenin may provide neuroprotection by regulating the levels of cytokines. Previous studies have shown that apigenin has the most anti-inflammatory effect compared to other structurally related flavonoids. 56,57 It has been demonstrated that apigenin inhibits the production of pro-inflammatory cytokines such as TNF-α and IL-6 through NF-κB signalling pathway. 19,20 NF-κB is a transcription factor that plays a central role in various cellular processes such as inflammation. 58 It has been demonstrated that production of inflammatory mediators is mainly regulated by NF-κB, and NF-κB binding sites in their promoters serve as inducible transcriptional regulatory elements. 59 Here, we demonstrated that apigenin reduces insulin fibril-induced NF-κB expression and this might account for reduction in NO production and pro-inflammatory cytokine gene expression (Figure 6(b)).
In summary, we have demonstrated that human insulin fibril is involved in oxidative stress, NO production, pro-inflammatory cytokines expression, and apoptosis among the affected SK-N-MC neuroblastoma cells. These observations are in agreement with the previous experimental findings obtained with disease-related proteins such as β-amyloid and α-synuclein on the same or different cell types. 8,10,43,44 In addition, our data have indicated that apigenin, as a multifunctional agent, is capable of protecting SK-N-MC cells against insulin amyloid-induced cytotoxicity (Figure 6(b)) mainly through pronounced modulation of the expression levels of inflammatory cytokines such as TNF-α and IL-6. Overall, these results provided the basis for introducing new generation of small organic molecules with dual function of free radical scavenging activity and suppressive effect on pro-inflammatory genes expression as their main mode of action.
Footnotes
Conflict of interest
The authors declared no conflicts of interest.
Funding
The authors thank the Research Council of University of Tehran for the financial support of this investigation.