
Abstract
Methyl bromide (MeBr) is a chemically reactive compound that has found use as a fire retardant and fumigant used for wood, soil, fruits and grains. Its use is banned in many countries because of its ozone-depleting properties. Despite this ban, the use of MeBr persists in some parts of the world (e.g. New Zealand) due to its important role in maintaining strict biosecurity of exported and imported products. Its high chemical reactivity leads to a broad toxicological profile ranging from acute respiratory toxicity following inhalation exposure, through carcinogenicity to neurotoxicty. In this article, we discuss the chemistry of MeBr in the context of its mechanisms of toxicity. The chemical reactivity of MeBr clearly underlies its toxicity. Bromine (Br) is electronegative and a good leaving group; the δ+ carbon thus facilitates electrophilic methylation of biological molecules including glutathione (GSH) via its δ− sulphur atom, leading to downstream effects due to GSH depletion. DNA alkylation, either directly by MeBr or indirectly due to reduction in GSH-mediated detoxification of reactive alkylating chemical species, might explain the carcinogenicity of MeBr. The neurotoxicity of MeBr is much more difficult to understand, but we speculate that methyl phosphates formed in cells might contribute to its neurone-specific toxicity via cholinesterase inhibition. Finally, evidence reviewed shows that it is unlikely for Br− liberated by the metabolism of MeBr to have any toxicological effect because the Br− dose is very low.
Introduction
Uses of methyl bromide
Methyl bromide (MeBr; bromomethane) is a heavy (density = 1.73 g cm−3) colourless gas (boiling point = 4.5°C) with a sweet chloroform-like odour at very high concentrations. It was introduced in the late 1800s as a fire suppressant; its high density was useful in this respect because it blanketed the fire and excluded oxygen. It was first used in fire extinguishers in the 1920s. It soon became clear that MeBr’s toxicity was not commensurate with its use in fire extinguishers. Indeed, as early as 1931, the British Physiologist, Sir Leonard Hill (1858–1952) described the toxic effects of MeBr following a series of experiments on himself; 1 this, in conjunction with cases of fatal poisoning following the use of MeBr to extinguish fires, 2 led to its fall from popularity. It was eventually withdrawn from use in fire extinguishers in the 1960s.
Despite its withdrawal from use as a fire retardant on the ground of unacceptable toxicity, this high toxicity profile supported its use as an insecticide. MeBr made its debut as an insecticide for use on grains in 1932. 3 Since then, it has found application in the eradication of bed bugs (Cimex lectularius), 4 treatment of lice 5 and nematodes 6,7 and the fumigation of food, 8 wood 9 and soil. 10,11
MeBr has also been used as a refrigerant 5 because MeBr gas is compressible and on decompression its latent heat results in temperature reduction – it was the precursor of chlorofluorocarbons in the refrigeration industry. Its use as a refrigerant steadily declined until its withdrawal in the 1950s.
Finally, MeBr found use in industrial processes as a methylating agent, for example, the methylation of 2-thiobarbituric acid to form 2-methylthiobarbituric acid and toluene to form xylene.
Ozone depletion and the Montreal Protocol
Most of the uses of MeBr involve the gas eventually being vented to the atmosphere; this has led to significant environmental problems associated with its chemical reactivity. For example, MeBr reacts in the stratosphere to liberate atomic bromine (Br), which in turn reacts with ozone to form oxygen and bromine oxide, thus depleting ozone (Figure 1). MeBr is now recognised as a major ozone depleting gas in the stratosphere and, under the Montreal Protocol (1992), its use was to be phased out by 2005 in the developed world and later by 2015 in developing countries (e.g. Chile). However, some countries (e.g. New Zealand), despite signing the Montreal Protocol, still use MeBr for export fumigation; indeed, a review of the use of MeBr carried out by the New Zealand Environmental Risk Management Agency in 2010 allows the continued import and use of MeBr in New Zealand with gas capture being introduced by 2020. 12 Therefore, the phase out has not been universally successful and the spirit of the Montreal Protocol has been lost.

Depletion of ozone by methyl bromide in the upper atmosphere showing the formation of bromine oxide, which results in further ozone depletion.
Toxic effects of MeBr in humans following occupational exposure
The first reported case of MeBr poisoning was in 1893, 13 when a man working with MeBr tried to transfer the liquid from one container to another using a pipe – to start the transfer, the worker sucked on the pipe. Within a short time the man became dizzy, had double vision, had difficulty in breathing and began to vomit. These symptoms continued for 4 days, and the man was admitted to a hospital where he suffered fits. He was discharged from hospital after 1.5 months and returned to work, but still complained of dizziness. This early case of MeBr toxicity displayed the, now well established, symptoms of MeBr poisoning; that is, an acute toxicity phase followed by a period of chronic toxicity that can persist for a long-time if the initial dose was high enough.
Recent reports of MeBr occupational exposure show a remarkably similar toxicity profile to the above 19th century case. In Japan, exposure of workers to termite fumigation 14,15 caused nausea, vomiting, dizziness, tremors and convulsions (one exposed worker died of heart failure). It soon became clear that the symptoms associated with acute exposure to MeBr fall into two distinct categories: local effects (e.g. nausea) and neurological effects (e.g. tremors).
There are few reports of low-dose chronic exposure to MeBr; however, case reports of chronically exposed workers showed electroencephalogram (EEG) abnormalities, 16 peripheral neuropathy 17 and deficits in the Wechsler Memory Scale. 18 Recently, In Port Nelson, New Zealand, the use of MeBr as a wood fumigant has been linked to motor neurone disease in port workers; 19,20 however, this remains controversial.
The chemical reactivity of methyl halides
MeBr is a simple alkyl halide with an electronegative Br attached to an electropositive carbon, which means that the molecule is polar (Figure 2) with δ+ carbon and δ− Br atoms. These charges determine the chemical reactivity of the molecule.
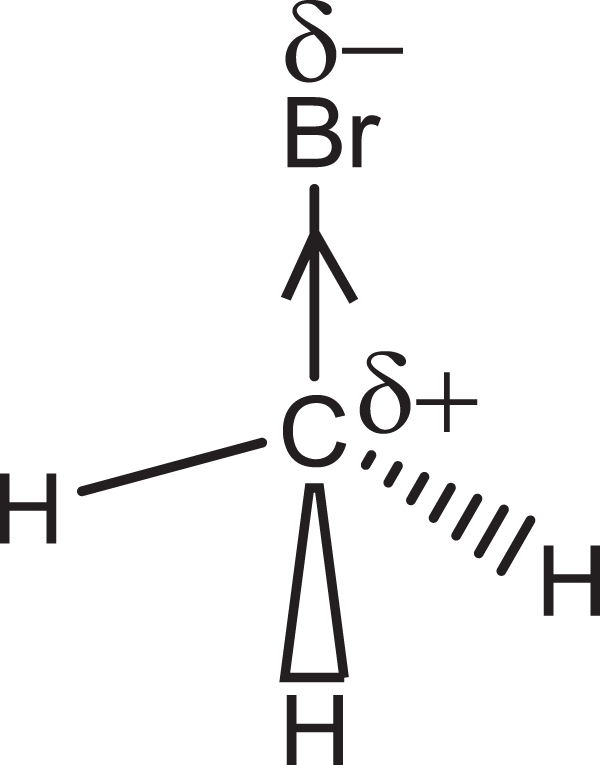
The molecular structure of methyl bromide showing the δ-charged bromine and carbon atoms.
The chemical reactivity of the alkyl halide series clearly shows that the larger the halogen (therefore, the lower its electronegativity), the greater the reactivity of the alkyl halide. This is because the larger halides are better leaving groups and so facilitate the formation of a carbonium ion (+CH3), which is a potent electrophile. This mechanism is well illustrated by methyl iodide (MeI) (Figure 3). Br is more electronegative than iodine, and therefore, it is unlikely that MeBr will form a carbonium ion. Continuing this line of reasoning, it is very likely that methyl chloride does not dissociate to form +CH3 and Cl− because of the extreme electronegativity of chlorine and the resulting short length and consequent strength of the C–Cl bond (Table 1).

Formation of a carbonium ion and iodide by dissociation of methyl iodide.
Electronegativities of the halogens, alkyl halide bond lengthsa and bond dissociation energies.

aRefer to bond lengths at http://www.science.uwaterloo.ca/~cchieh/cact/c120/bondel.html
MeBr does not dissociate to form a carbonium ion during its reactions with nucleophiles; instead, it forms a transition state (Figure 4). On the other hand, MeI does dissociate to a carbonium ion and iodide. Therefore, MeBr is far less reactive in biological systems than MeI, which is well illustrated by the different hydrolysis rates of the alkyl halides to form methanol and the corresponding hydrogen halide (Figure 5).
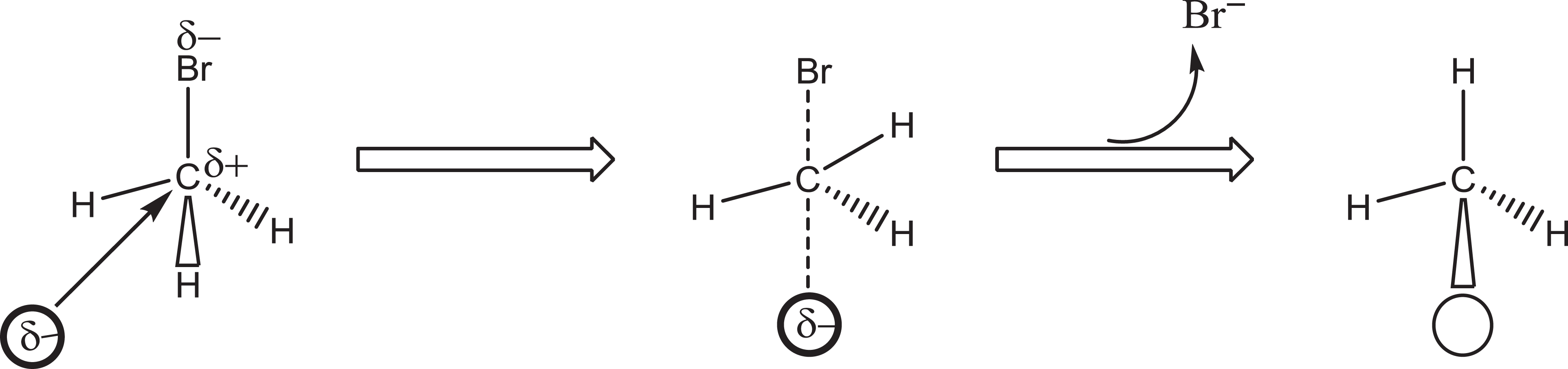
A nucleophile (δ−) attacking the δ+ carbon of methyl bromide to form an unstable transition complex, which breaks down to form the reaction product and bromide.

The hydrolysis of alkyl halides to form the corresponding alcohol and halide (X) ion (top) and the specific hydrolysis reaction for methyl bromide showing the formation of methanol and bromide (bottom).
The hydrolysis rate constants (298 K) for MeBr and MeI are 3.57 × 10−7 s−1 and 6.87 × 10−8 s−1 respectively, 21 showing that MeI hydrolyses much faster than MeBr. This general principle would also apply to other reactions of the positively charged carbon (i.e. a δ+ carbon for MeBr and carbonium ion for MeI) of the alkyl halide molecule (Figures 3 and 4). For example, methylation of nucleic acid bases in biological systems (e.g. adenine; Figure 6).

Methylation of adenine by methyl bromide to form 3-N-methyl adenine.
Polarity and reactivity of MeBr: Implications for transport and residence time in biological systems
MeBr is slightly water soluble (solubility at 20°C = 1.75%), 22 which reflects its low molecular polarity (Figure 2). The dipole moment of MeBr is 1.8203D 23 (D is a measure of the distance between polar centres; 1 = minimum separation, 11 = maximum separation), which means that it has relatively low polarity. Even though MeBr is only slightly water soluble, in a biological context, its solubility is likely to allow it to be transported in the circulatory system and within cells at toxicologically significant concentrations.
Despite water solubility of MeBr in an in vivo toxicological context, its low dipole moment means that it is also relatively hydrophobic; log POW [20°C] = 1.19, 24 and therefore, it would be sufficiently soluble in the lipid component of biological membranes to allow it to be taken up by cells but not soluble enough to accumulate. 25
Therefore, MeBr’s polarity facilitates its uptake by and transport in biological systems. However, its chemical reactivity limits its residence time in biological systems, where there are myriad molecules with which MeBr can react. This is illustrated well in studies on rats, in which MeBr was rapidly eliminated from rat tissues (with half-life (t ½) of approximately 30 min) with Br− being excreted in urine and faeces (t ½ = 9.6 h and 16.1 h, respectively 26 ). The fate and behaviour of MeBr in rats illustrate the importance of chemical reactivity; it is likely that the chemistry underpinning the pharmacokinetics involves hydrolysis of MeBr to liberate Br− (Figure 4).
This leads to rapid clearance of MeBr from tissues with concomitant excretion of Br− as seen in the rat studies discussed above.
Metabolism and excretion of MeBr
Animal studies have shown that MeBr is well absorbed following inhalation exposure and is excreted mainly as Br− and carbon dioxide (CO2). The excretion t ½ of Br− following inhalation exposure of rats to MeBr is approximately 5 days; 26 however, the t ½ of exhalation of CO2 (from oxidation of MeBr’s methyl group) is at least biphasic and very much shorter (3.9–11.4 h 27 ) than the excretion t ½ of Br−. The t ½ of CO2 and Br− suggest a multi-pathway metabolism that involves degradation of MeBr to form methanol (reaction of MeBr with water is likely to be spontaneous) with concomitant elimination of Br−. The methanol is then oxidised to formate, which enters the 1-carbon metabolic pool. A proportion of the formate is oxidised to CO2 + H2O, which explains the rapid elimination of CO2 following MeBr administration. The remaining formate is metabolised via oxalate and the tricarboxylic acid cycle to form amino acids (including homocysteine and cysteine 28 ). Interestingly, studies administering oral dose of [14C]-MeBr in rats showed elimination of approximately 46% of the administered radioactivity in bile within 24 h of dosing. Since only large molecules are excreted in bile – in the rat, the excretion threshold is 325 ± 50 Da 29 – this suggests that the 14CH3-group originating from the MeBr molecule has been incorporated into a molecule of molecular weight sufficient to allow it to be excreted in bile. MeBr is thought to methylate glutathione (GSH) in vivo; indeed, this is an important facet of the mechanism of toxicity of MeBr. 19,30 It is possible that the [14C] molecule excreted in bile following [14C]-MeBr administration is either [14 C]-methylglutathione or one of its degradation products (e.g. [14C]-methyl-cysteinylglycine). In addition, methylglutathione is metabolised to formaldehyde via S-methylcysteine, methylthioacetic acid and methanethiol, 31 and formaldehyde is oxidised in cells to formate, 32 which provides another metabolic route for the MeBr methyl group to enter the cellular 1-carbon pool (Figure 7).

A proposed metabolic pathway for methyl bromide showing routes of excretion.
Neuropathology following human exposure to MeBr
Pathological changes following exposure to a chemical often indicate the anatomical region of the chemical’s toxic effects. Since exposure to MeBr causes neurological symptoms, there have been a plethora of reports of neuropathological changes in occupationally MeBr-exposed humans, including:
Brain magnetic resonance imaging (MRI) findings: MRI of MeBr-exposed people has shown strikingly symmetric brainstem and cerebellar lesions. These patients’ clinical course and the topography and resolution of MRI abnormalities suggest that MeBr exposure results in energy deprivation syndrome. 24
Electron microscopical studies of neurones: Electron microscopy of neurological tissue from MeBr-exposed people showed degeneration of both myelinated and unmyelinated fibres. In these samples, numerous Schwann cell subunits were devoid of axons and showed segregated axonal microtubules. 33
EEG disturbances: Abnormal EEG patterns, consistent with the early stages of encephalopathy, have been found in MeBr-exposed people. 15
From the above findings, it is clear that following human exposure to MeBr, neurological changes develop that are likely to underpin the neurological symptoms seen in occupationally MeBr-exposed people.
Neurophysiological changes following human exposure to MeBr
Neurophysiological changes following exposure to a chemical give information about the microanatomical region of toxic effects. The following neurophysiological studies in people exposed to MeBr have been reported:
Nerve conduction tests: Nerve conduction tests have been carried out on MeBr-exposed fumigation workers to detect peripheral neuropathy. The results showed significantly reduced performances on the Pattern Memory test, Santa Ana Dexterity test of the dominant hand and olfactory testing. 34 In addition, F-wave latencies were long when the peroneal motor nerve was tested in MeBr-exposed people; 34 this suggests that a slow motor response results following exposure to MeBr.
Muscle action potential amplitude measurements: Tibial and peroneal nerves showed reduced motor conduction and muscle action potential amplitudes in MeBr-exposed people compared to controls. 33
Clearly, the pathological changes that have been shown following human exposure to MeBr translate into neurophysiological effects that, in turn, might underlie some of the symptoms that result following exposure (both acute and chronic) to MeBr (e.g. peripheral neuropathy).
Biochemical changes following human exposure to MeBr
Biochemical changes after exposure to a chemical might point to specific mechanisms of toxicity. There have been several studies published on occupationally MeBr-exposed humans that report biochemical parameters post exposure, including:
Blood [Br−]: Serum [Br−] was significantly higher in MeBr-exposed agricultural workers (mean ± SD = 15.33 ± 1.90 mg L− 1) than in controls (mean ± SD = 4.13 ± 1.05 mg L− 1). 35 This is an indicator of exposure to MeBr and presents evidence for debromination as an important pathway of MeBr metabolism or, perhaps, alkylation (with subsequent loss of Br−) as a mechanism of its toxicity. Br− per se is not thought to cause the neurotoxicity associated with MeBr exposure. 36
Cysteine adducts (e.g. S-methylcysteine) in blood: S-Methylcysteine has been shown to be present in the circulatory systems of rodents and humans exposed to MeBr. 37 It has been suggested that the methylcysteine formed might be neurotoxic due to its structural analogy (Figure 8) with γ-aminobutanoic acid (GABA) and possible interaction with GABA receptors. 36 Indeed, rat hippocampal slices exposed to S-methylcysteine (10− 2 M for 30 min) had reduced synaptically evoked population spikes in the Cornu Ammonis (CA1) pyramidal cell region, thus suggesting a neurotoxic response to S-methylcysteine.
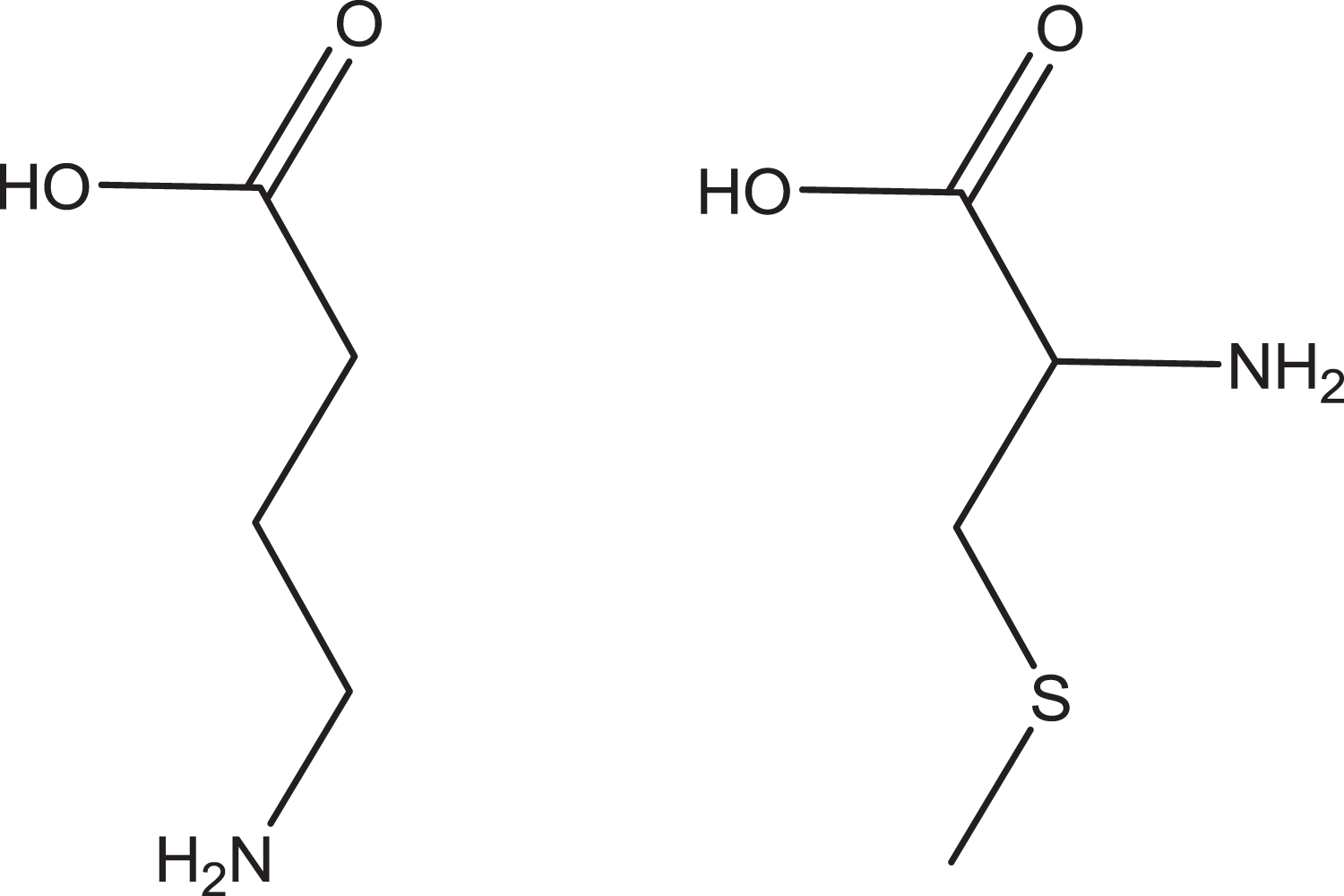
Molecular structures of GABA (left) and S-methylcysteine (right) – their structural analogies might mean that S-methylcysteine can interact with the GABA receptor. GABA: γ-aminobutanoic acid.
Mechanisms of MeBr toxicity
Mutagenicity/carcinogenicity
Methylation of single-stranded nucleic acid bases by alkyl halides leads to many possible O- and N-methyl derivatives, including 7-methylguanine, 3-methyladenine and 3-methylcytosine. 38 These alkylation mutations are important in the mechanism of carcinogenesis of the alkyl halides because they lead to DNA repair damage and/or misreading of the nucleic acid template. The reactivity of the particular alkyl halide determines the degree of nucleic acid methylation that will occur in vivo following exposure. One would expect MeI to be a far more effective biological methylating agent than MeBr; however, both MeI and MeBr are both mutagenic in the Salmonella typhimurium His− reversion assay (Ames’ Test) with and without a metabolising system (S9 mix) 39,40 and both are carcinogenic in animals. 41,42,43 Interestingly, workers exposed to MeBr show indicators of genotoxicity (e.g. micronuclei in lymphocytes). Rats exposed to [14C]-MeBr form 3-N-[14C]-methyl adenine and 7-N-[14C]-methyl- and/or 6-O-[14C]-methyl guanine adducts, 44 which illustrate the methylating properties of MeBr in a cellular context and points to methylation as a mechanism of mutagenicity and/or carcinogenicity of MeI and MeBr in vivo.
Based on MeBr’s chemistry and its in vivo effects, it is clearly reactive in a biochemical context by virtue of its δ+ carbon atom. This level of biochemical reactivity means that it will attack only electron rich (i.e. strongly δ−) atoms in biological molecules. For example, in our laboratory, using nuclear magnetic resonance spectroscopy, we have shown , that MeBr methylates GSH thus depleting the cellular GSH pool 19 and that the reaction rate is increased by glutathione-S-transferase but does proceed uncatalysed (Bulathsinghala and Shaw, 2012).
Neurotoxicity
It is well known that occupational exposure to MeBr causes broad symptoms of neurotoxicity (e.g. tremors; see discussion above and Clarke et al.). 2 However, the underlying mechanisms of these neurological symptoms are not understood.
Interestingly, it is possible that MeBr methylates phosphate to form monomethyl phosphates and dimethyl phosphates (Figure 9). Methyl phosphates (MPs) are the simplest organophosphates (OPs; Figure 10). These simple OPs do not have a good leaving group; they, therefore, are thought not to be acetylcholinesterase (AChE) inhibitors; however, MPs are known to react with water to form methanol and phosphoric acid, which demonstrates that MPs reacts with the –OH of water and suggests that they might also react with other –OH groups. 45 The mechanism of inhibition of AChE by OPs is via phosphorylation or alkyl phosphorylation of an active site serine –OH; therefore, it is possible that MPs would also alkyl phosphorylate the AChE active site serine and so inhibit the enzyme. The degree of inhibition depends on the magnitude of serine phosphorylation. It is interesting to speculate that if MeBr methylates, cellular phosphates and MPs are AChE inhibitors that might explain the neurological specificity of MeBr’s toxicity, especially following prolonged low doses. We must emphasise that this hypothesis is speculation, but perhaps should be considered in light of AChE inhibition by methyl parathion. 46
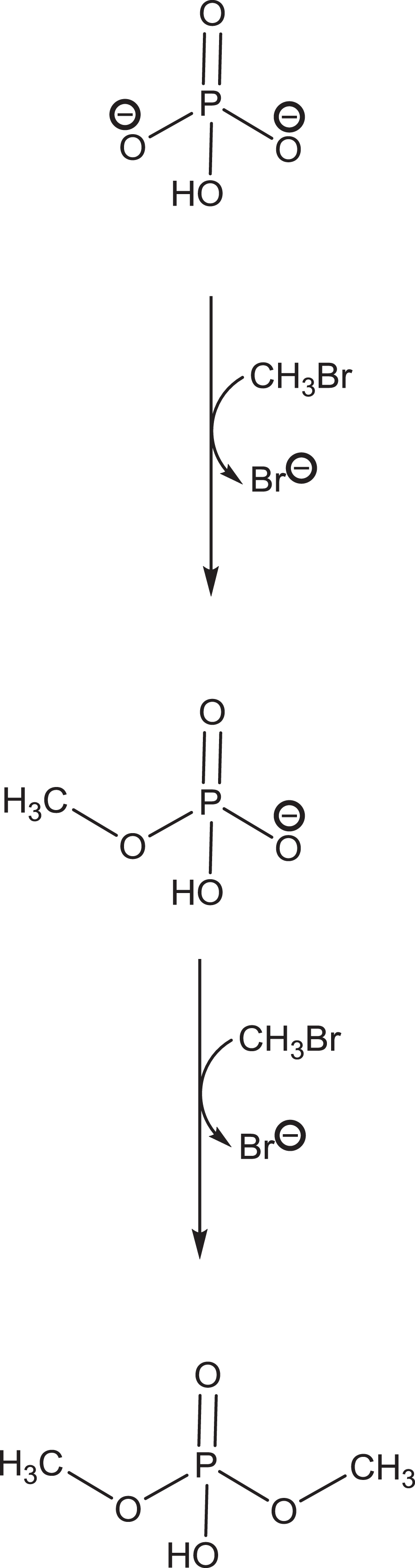
The formation of mono- and dimethyl phosphates when methyl bromide is incubated under simulated cellular conditions.

Comparision of (a) methylphosphate with (b) sarin and (c) diazinon, the two potent AChEs inhibiting OPs. AChE: acetylchlinesterase; OPs: organo phosphates.
The symptoms of OP poisoning following low dose, multiple exposures are diverse and include many symptoms associated with AChE inhibition (e.g. salivation and tremors). 47 Interestingly, the reported symptoms of MeBr intoxication are nausea, vomiting, dizziness, tremors and convulsions; 24 all these symptoms are also associated with OP exposure and the latter three, in particular, could be explained by AChE inhibition, which again suggests that MeBr might have an AChE inhibition-based mechanism of toxicity.
Bromide toxicity
Ingestion of MeBr results in the metabolic liberation of Br−. It is therefore important to consider whether bromide might, at least in part, explain the toxicity of MeBr. Br− is toxic only at very high doses (median lethal dose (rat, oral) = 3500–7000 mg kg−1 body weight 48 ), and it is highly unlikely that such doses would be attained by metabolic liberation following occupational exposure to MeBr.
The symptoms of Br− intoxication following the clinical use of potassium bromide (KBr) are very varied, including skin lesions, psychoses, headaches, fatigue, sleeplessness, dizziness, disorientation, tremors and unsteady gait. While some of these symptoms might be explained by neurological interactions (e.g. dizziness and tremors), the KBr doses used clinically lead to blood bromide concentrations of approximately 500 mg L−1,49 which is far in excess of the blood bromide levels (e.g. 15 mg L−1) 35 found following occupational exposure to doses of MeBr that resulted in symptoms of toxicity. In addition, Br− is quickly excreted in urine and therefore is not cumulative in a toxicological context. Therefore, it is very unlikely that the toxicity of MeBr is in any way related to the toxic effects of bromide, even following prolonged low-dose exposure to MeBr.
Distribution of MeBr following exposure
Studies in rats administered with [14C]-MeBr doses have shown that radioactivity is well distributed around the animal’s body and is not concentrated in neurological tissue. 49 This is interesting because it suggests that the observed neurotoxic effects following exposure to MeBr are not due to MeBr or its metabolites interacting with neurological tissue in such a way that requires their concentrations in nervous tissue. In view of this, the only possible mechanism of neurotoxicity would involve transient biochemical effects (e.g. inhibition of AChE). This, of course, is speculation, but is an interesting possibility because the chemical reactivity of MeBr and its consequent cellular biochemistry point to such a mechanism of toxicity. The finding that MeBr, or its metabolites, is not concentrated in neurological tissues and is at lower concentrations than in other tissues in rats administered with MeBr dose. 50 For example, Jascok et al. 50 demonstrated that following inhalation exposure of rats to [14C]-MeBr that their brains contained only 8% of the radioactivity present in their liver and that brain radioactivity was approaching background levels 32 h after dosing. 50 This supports the hypothesis that MeBr alters neurological cell biochemistry in such a way as to cause long-term harm without the MeBr molecule being present for more time than is necessary to initiate the biochemical changes. This supports the GSH depletion hypothesis discussed above.
Conclusions
MeBr is very toxic to animals, including humans, and there have been numerous accounts of occupational exposure leading to severe neurotoxicity. In view of this, its symptoms of toxicity are well documented. MeBr has high chemical reactivity on account of the molecular polarity induced by its electronegative Br with the concomitant formation of a δ+ carbon. This high chemical reactivity almost certainly explains its high toxicity due to its reaction with important cellular components. In addition, MeBr’s high chemical reactivity explains its ozone depleting properties. As a result of its toxicity and ozone depletion, the use of MeBr is being phased out in most countries. While the chemical reactivity of MeBr, in particular its catalysis of ozone depletion, is well understood, surprisingly, its mechanisms of toxicity remain obscure.
Following our review on the chemical reactivity of MeBr, published studies in animals and observations in MeBr-poisoned humans, we speculate that reaction of MeBr with GSH, resulting in GSH depletion, might explain its general cellular toxicity. However, we feel it is unlikely that GSH depletion fully explains MeBr’s neurotoxicity, especially as MeBr does not appear to accumulate in neurological tissues. We also speculate that the formation of MPs in vivo and the possibility that they might inhibit AChE could, at least in part, explain the neurotoxicity of MeBr – this is the subject of further study in our laboratory.
Finally, bromide liberated from MeBr in cells is very unlikely to have a role in MeBr’s mechanism of toxicity because cellular bromide concentrations are unlikely to reach toxic levels, but it is possible that S-methylcysteine (produced by reaction between MeBr and cysteine) might contribute to MeBr’s mechanism of neurotoxicity via its interaction with the GABA receptor.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.