
Abstract
Mitogen-activated protein kinases (MAPKs) are involved in neuronal death caused by many cytotoxins. Conventional MAPKs consist of three family members: extracellular signal-regulated kinase-1/2 (ERK1/2), c-Jun N-terminal kinase (JNK) and p38. It has been originally shown that ERK1/2 is important for cell survival, whereas JNK and p38 are deemed stress responsive and thus involved in apoptosis. However, information describing the role of MAPKs in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced neurotoxicity is insufficient. The aim of this study was to identify the role of MAPK cascades in TCDD-induced neurotoxicity using differentiated pheochromocytoma (PC12) cells as a model for neuronal cells. Cell viability assay, terminal deoxynucleotidyl transferase dUTP nick-end labeling assay and flow cytometry analysis showed that TCDD attenuated cell viability with a dose- and time-dependent manner and significantly induced apoptosis in primary cortical neurons and PC12 cells. Western blot analysis indicated that TCDD markedly activated the expression of ERK1/2, JNK and p38 in TCDD-treated PC12 cells. Furthermore, PD98059 (ERK1/2 inhibitor), SP600125 (JNK inhibitor) and SB202190 (p38 inhibitor) notably blocked the effect of TCDD on cell apoptosis. Based on the findings above, it is concluded that the activation of MAPK signaling pathways may be associated with TCDD-mediated neuronal apoptosis.
Introduction
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is a prototypical environmental contaminant that is considered to be associated with teratogenic effects in the immune and reproductive systems 1–3 and can cause neurobehavioral abnormalities associated with both cognitive and locomotor systems. 4,5 Aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, plays an important role in the regulation of toxic and carcinogenic properties of TCDD. 6–8 Additionally, TCDD-bioactivated fetal enzymes like the cytochromes P450 (CYPs) can initiate reactive oxygen species (ROS) accumulation, resulting in oxidative damage to altered signal transduction, which can regulate cell cycle and apoptosis in many cell lines. 9,10 Recent studies have demonstrated that TCDD can target GABAergic neurons in rat brain, and these neurons in turn mediate developmental effects of this contaminant on the reproductive function. 11,12 In the dorsal midbrain of zebrafish embryos, a predictive model for assessing neurotoxicity, TCDD has been reported to cause apoptosis by activation of AhR. 13 In particular, an epidemiological study by Rogan and Gladen suggested that children who are accidentally exposed to TCDD-related molecules of polychlorinated biphenyls exhibit delayed motor development and higher incidences of hypotonia. 14 In conclusion, TCDD has prominent neurotoxicity on nervous systems, and the AhR receptor is essential in its cytotoxin release. However, the underlying mechanisms of TCDD-induced neurotoxicity still remain to be perfected.
Mitogen-activated protein kinases (MAPKs), including extracelluar signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK) and p38, are involved in neuronal apoptosis caused by various stimuli.
15–17
MAPKs are an important group of serine/threonine kinases that participate in cell differentiation, cell growth and survival. Phosphorylation of ERK1/2 promotes cell survival, whereas activation of JNK and p38 induces apoptosis in response to multiple “stress” stimuli.
18
Previous studies on the role of individual MAPKs in the TCDD-induced apoptosis indicated that the activation of MAPK pathways is different to cell types. For example, in the RAW 264.7 cells, activation of p38 plays a critical role in the apoptosis induced by TCDD,
19
while JNK and ERK1/2 are activated in TCDD-induced cell apoptosis in Jurkat
Primary cortical neurons from late embryonic brain tissue rapidly extend neurites when prepared in a dissociated culture and develop distinct axons and dendrites consistent with the morphology of neurons observed in the brain in vivo. 21 The pheochromocytoma (PC12) cell line is derived from a PC12 of the rat adrenal medulla and can be differentiated into a neuronal phenotype by stimulation with nerve growth factor (NGF). NGF-treated PC12 cells stop dividing, develop neurites, display electrical activity and develop many other properties similar to those of sympathetic neurons. 22,23 Primary cortical neurons and PC12 cells are both considered to be useful models for neurobiology research. In particular, 4,6-diamidino-2-phenylindole (DAPI) and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining instead of flow cytometry (FCM) analysis were used to detect the apoptosis of primary cortical neurons due to the extensive axonal and dendritical connections established between neurons, while the apoptosis of PC12 cells can be detected by FCM analysis.
In conclusion, MAPKs have been reported to play an important role in neuron death caused by many cytotoxins; however, what role MAPKs play in TCDD-induced neurotoxicity remains unclear. Therefore, the present study was performed to evaluate the role of MAPK cascades in TCDD-induced neurotoxicity using differentiated PC12 cells as a model for neuronal cells.
Materials and methods
Primary culture and treatment
Primary cultures of cortical neurons were prepared from the brain of E18–E19 Sprague-Dawley rat embryos obtained from the Experimental Animal Center of Nantong University, China. In brief, rat embryos (n = 8–10) were killed by cervical dislocation under anesthesia, their brains were quickly removed and the cortexes were harvested on a cold stage, then cortical tissues were dissected out and treated with 0.25% trypsin for 15 min at 37°C in calcium (Ca2+) and magnesium ion (Mg2+)-free Hank’s balanced salt solution, the cortex were washed in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) to stop trypsin activity. Then all obtained primary cells were mixed and resuspended in DMEM supplemented with 10% FBS and plated onto poly-
The primary cultured cortical neurons were treated for 24 h with or without different concentrations of TCDD (0.1, 1, 10, 20, 50 or 100 nM), or treated with or without 50 nM TCDD for different time (0.5, 1, 3, 6, 12 or 24 h) at 37°C in a humidified atmosphere of 95% air and 5% CO2. The cortical neurons cultured in `plain medium served as the control group. At the end of cell treatments, different tests were carried out as described below.
PC12 cell culture and treatment
Differentiated PC12 cells were cultured in DMEM with 2 mM glutamine (Sigma, St Louis, MO, USA), 100 units/ml penicillin and 100 μg/ml streptomycin with 10% FBS (Hyclone, Logan, UT, USA) at 37°C under 5% CO2 in humidified air. In order to study the cytotoxicity induced by TCDD, the cells were treated for 24 h with or without different concentrations of TCDD (1, 10, 100, 200, 500 or 1000 nM) or treated with or without 200 nM TCDD for different time (0.5, 1, 3, 6, 12 or 24 h). To further elucidate the role of ERK1/2 or JNK or p38 in TCDD-induced apoptosis, the cells were pretreated with PD98059 (ERK inhibitor) or SP600125 (JNK inhibitor) or SB202190 (p38 inhibitor) for 60 min before exposure to TCDD.
Cell viability assay
Primary cortical neurons and PC12 cells were plated at a density of 5 × 104 cells/well in 96-well plates, and the cell viability was assessed by the Cell Counter Kit-8 (CCK-8) assay (Dojindo Laboratories, Kyushu, Japan). Briefly, the cells were incubated at 37°C with the indicated concentrations of TCDD for 24 h. After the indicated treatments, CCK-8 solution of 10 μl was added to each well of the plate and the cells in the plate were incubated for 2 h in the incubator, and the absorbance was quantified on an automated reader (Bio-tek, Hartford, VT USA). Cell viability of time course experiments was also measured using CCK-8 kit according to the manufacturer’s instruction.
TUNEL assay
The TUNEL assay using the In Situ Cell Death Detection Kit (Roche Applied Science, Mannheim, Germany) was used to confirm apoptosis by demonstrating apoptotic bodies in primary cortical neurons. Briefly, after exposure to TCDD for 24 h, primary cortical neurons were fixed with 4% formaldehyde and incubated at room temperature for 40 min. Neurons were washed with phosphate-buffered solution (PBS) and then permeabilized in Triton
FCM analysis of apoptosis
Annexin-V-FLUOS Staining Kit (Roche Diagnostics GmbH, Penzberg, Germany) was used following the protocol recommended by the manufacturer. PC12 cells treated with or without TCDD (100 or 200 nM) for 24 h were harvested and resuspended at a concentration of 1 × 106 cells/ml in 100 µl of Annexin-V-FLUOS labeling solution containing 20 µl of the Annexin-V-FLOUS solution with 1000 µl of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (10 mM HEPES/sodium hydroxide, pH 7.4, 140 mM sodium chloride, 5 mM calcium chloride) and supplemented with 20 µl of propidium iodide (PI; 50 µg/ml). After 15 min incubation in the dark at room temperature, cells were analyzed within 1 h on a flow cytometer (BD FACScalibur, BD Bioscience, San Jose, California, USA)
DAPI staining
The fluorescent dye DAPI was used to detect nuclear fragmentation, which is a characteristic of apoptotic cells. Primary neuronal cultures (5 × 105 cells/well in 24 well plates) were incubated with 50 nM TCDD for 24 h and then washed with PBS and fixed with 4% paraformaldehyde for 40 min at room temperature. The fixed cells were then washed with PBS and stained with DAPI. Following 10 min of incubation, the cells were again washed with PBS and the plates were observed under a fluorescence microscope.
Western blot analysis
After subjected to the indicated treatments, approximately 5 × 106 cells were washed with cold PBS for three times and lysed with cell lysis solution. Sample buffer was added to cytosolic extracts, and after boiling for 10 min, equal amounts of supernatant from each sample were fractionated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrotransferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% skim milk for 2 h at room temperature. Then the membranes were incubated with the antibody for caspase-3, phosphorylated-ERK1/2 (p-ERK1/2) or ERK1/2, phosphorylated-JNK (p-JNK) or JNK and phosphorylated-p38 (p-p38) or p38 (1:1000; cell signaling) at 4°C overnight. At last, immunoreactive bands were then detected by incubation with conjugates of anti-rabbit immunoglobulin G with horseradish peroxidase (1:2000; Southern-Biotech) for 2 h and visualized using enhanced chemiluminescence system (Pierce Company, Woburn, MA, USA).
Statistical analysis
The data are expressed as mean ± SEM. Student’s t test was used to compare control group and TCDD-only treated group. One-way analysis of variance followed by Tukey’s multiple comparison test were used to analyze differences between groups. p < 0.05 was considered statistically significant. All data were analyzed with SPSS13.0 software.
Results
TCDD-induced apoptosis in primary cortical neurons
To analyze the cytotoxic effects of TCDD exposure on neurons, we first determined the cell viability in primary cortical neurons. After 24 h of TCDD exposure, cells treated with 1 nM and less TCDD maintained a high cell viability, whereas concentrations of 10 nM and greater produced a significant increase in cell death (p < 0.01, Figure 1(Aa)). At the same time, 50 nM TCDD was selected as the dose of time course experiments. Treatment with 50 nM TCDD for different times (0, 0.5, 1, 3, 6, 12 or 24 h) demonstrated that the cell viability was inhibited significantly at 6 h (p < 0.01, Figure 1(Ab)). These results show that the cell viability of the neurons can be effectively inhibited by TCDD in a dose- and time-dependent manner.

Apoptosis induced by TCDD in primary cortical neurons. (A) (a) Primary cortical neurons isolated from the brain of E18–E19 SD rat embryos were cultured for 7 days and then treated with various concentrations of TCDD for 24 h. (b) Primary cortical neurons were exposed to 50 nM TCDD for the indicated times and cell viability assessed at each time point, n = 6 wells for all CCK-8 assay. (B) Images a and d show representative cell morphologies, while images b and e show representative nuclear morphologies using DAPI staining with fluorescent microscopy. Images c and f display TUNEL-positive cells (green) following TUNEL/DAPI staining and all the nuclei stained with DAPI (blue). Neurons were cultured without TCDD (a, b and c) or 50 nM TCDD (d, e and f) for 24 h. White arrow shows the network of neurons expressing neurites in image a (white arrow) in comparison with image d. White arrow in (b) shows normal nuclei, while white arrow in (e) indicates apoptotic and fragmented nuclei. (C) The ratio of TUNEL-positive cells in the whole cell population (n = 6). (D) (a) Cell lysates were analyzed by Western blotting using anti-caspase-3 antibodies (top) and anti-GAPDH antibodies (bottom). (b) Densitometer scan of active caspase-3/GAPDH ratio. Values are indicated as the mean ± SEM. Statistical significance was determined as described in Materials and methods section. **p < 0.01; ***p < 0.001. TCDD: 2,3,7,8-tetrachlorodibenzo-p-dioxin; SD: Sprague-Dawley; CCK-8: Cell Counter Kit-8; DAPI: 4,6-Diamidino-2-phenylindole; TUNEL: terminal deoxynucleotidyl transferase dUTP nick-end labeling; GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
To further detect the role of TCDD on neuronal apoptosis, cellular and nuclear morphology were documented at 24 h following exposure to 0 or 50 nM TCDD in primary cortical neurons. Neurons in cultures treated with 50 nM TCDD had their dendritic processes reduced in number and length and the complete dendritic network significantly disappeared when compared with normal neurons (Figure 1(Ba) and (Bd)). The DAPI staining showed that neurons exposed to 50 nM TCDD exhibited strong chromatin condensation and apoptotic bodies, but it was not observed in normal neurons (Figures 1(Bb) and (Be)). In addition, TUNEL assay showed that a 50-nM TCDD exposure increased the number of TUNEL-positive neurons (21.1 ± 1.7%) compared with controls (5.0 ± 0.6%; p < 0.001, Figure 1(Bc) and (Bf)). Moreover, the active caspase-3 protein level of primary cortical neurons treated with 50 nM TCDD was found to be significantly increased after a 24-h exposure (Figure 1(C)) and the kinetic profile of the expression of active caspase-3 revealed a time-dependent manner in the presence of 50 nM TCDD (Figure 1(D)). All these results confirm that TCDD significantly induces apoptosis in primary cortical neurons.
TCDD-induced apoptosis in PC12 cells
To support our previous conclusions, we continued to study TCDD-mediated neurotoxicity using PC12 cells. A similar result was observed in TCDD-treated PC12 cells that TCDD produced a decrease in cell viability in a dose- and time-dependent manner (Figure 2(A)). The percentage of early apoptosis (Annexin-V positive/PI negative) and necrosis/late apoptosis (Annexin-V positive/PI positive) was increased in PC12 cells challenged with 100 or 200 nM TCDD for 24 h compared with the untreated cells (Figure 2(B)). Moreover, the expression of active caspase-3 began to increase as early as 0.5 h and peaked at 6–12 h (Figure 2(C)). In summary, these results suggest that TCDD also induces apoptosis in PC12 cells. PC12 cells were used to identify the TCDD effect on cell apoptosis in the following studies.

Apoptosis induced by TCDD in PC12 cells. (A) (a) PC12 cells were seeded into a 96-well flat-bottomed microplate and incubated with various concentrations of TCDD for 24 h. (b) Cells were seeded into 96-well flat-bottomed microplate and incubated with 200 nM TCDD for the indicated times, n = 6 wells for all CCK-8 assay. (B) PC12 cells were stained with Annexin-V and PI, followed by flow cytometry analysis. (C) (a) Cell lysates were analyzed by Western blotting using anti-caspase-3 antibodies (top) and anti-GAPDH antibodies (bottom). (b) Densitometer scan of active caspase-3/GAPDH ratio. Data analyzed with SPSS13.0 software are expressed as mean ± SEM (n = 3). **p < 0.01; ***p < 0.001. TCDD: 2,3,7,8-tetrachlorodibenzo-p-dioxin; PC12: pheochromocytoma; CCK-8: Cell Counter Kit-8; PI: propidium iodide; GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
Involvement of MAPKs in TCDD-induced apoptosis
To investigate whether the ERK1/2, JNK and p38 MAPK pathways are involved in TCDD-induced apoptosis in PC12 cells, the activation of MAPKs signal pathway was detected by Western blotting. As illustrated in Figure 3(A), the activation of p-ERK1/2 increased at 1 h and reached the peak at 6 h after TCDD treatment (p < 0.001); however, the total ERK1/2 expression in the indicated times had no change. At the same time, TCDD strongly provoked a rapid increase in the levels of activation of p-JNK, and both phosphorylated-p54 and phosphorylated-p46 JNKs with maximum activity peaked at 3 h (p < 0.001) before declined toward basal level within 24 h, and the expression of total JNK was also unchanged during exposure of cells to TCDD. In addition, TCDD significantly activated p-p38 MAPK, which was also maximal at 3 h (p < 0.001) and sustained for up to 12 h, but expression of total p38 MAPK decreased significantly at 12 and 24 h. These results clearly demonstrate that TCDD stimulates all three members of MAPKs in PC12 cells, but with differential kinetics of activation.
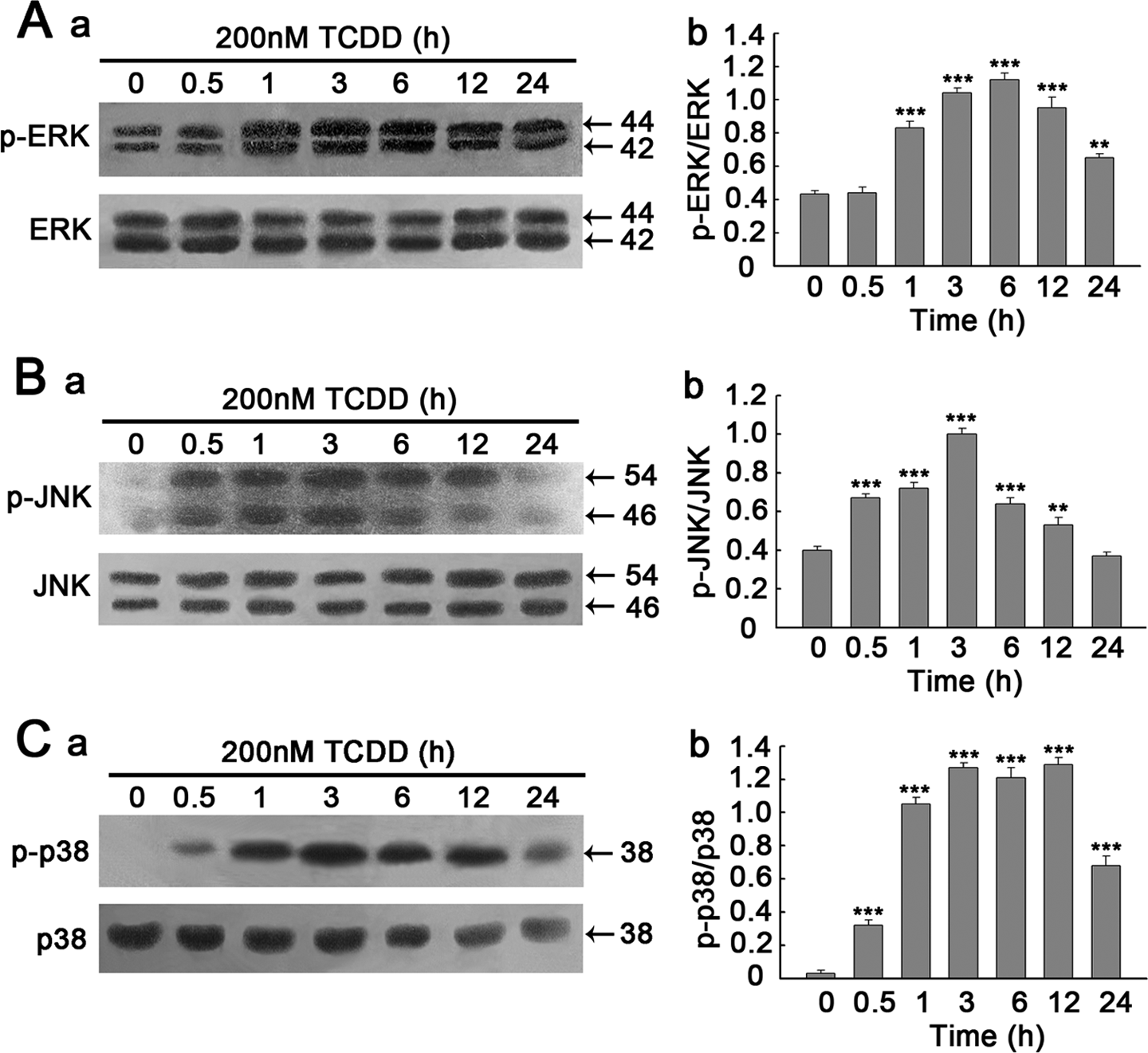
Time courses of TCDD-induced phosphorylation of MAPKs. PC12 cells were treated with 200 nM TCDD and harvested at the indicated times (0, 0.5, 1, 3, 6, 12 or 24 h). Whole-cell lysates (20 μg) were resolved by SDS-PAGE (10%) followed by Western blotting using a set of antibodies that recognized either activated or total MAPKs expression. (A) (a), (B) (a) and (C) (a) time courses for the effects of TCDD on phosphorylation of ERK1/2, JNK and p38, respectively. (A) (b), (B) (b) and (C) (b) Denstiometric analysis for the results in (A) (a), (B) (a) and (C) (a), respectively. Data are presented as the mean ± SEM (n = 3). **p < 0.01; ***p < 0.001 compared to the control group (0 h). TCDD: 2,3,7,8-tetrachlorodibenzo-p-dioxin; MAPKs: mitogen-activated protein kinases; PC12: pheochromocytoma; SDS-PAGE: sodium dodecyl sulfate–polyacrylamide gel electrophoresis; ERK1/2: extracellular signal-regulated kinase1/2; JNK: c-Jun N-terminal kinase.
Effects of PD98059, SP600125 and SB202190 on TCDD-induced MAPKs phosphorylation
To further verify the role of the ERK1/2, JNK and p38 MAPK pathways in TCDD-induced apoptosis in PC12 cells, PD98059 (ERK1/2 signal pathway inhibitor), SP600125 (JNK signal pathway inhibitor) and SB202190 (p38 signal pathway inhibitor) were added to PC12 cells. PC12 cells were pretreated with the PD98059, SP600125 and SB202190 at different concentrations for 1 h and then stimulated with 200 nM TCDD for 3 h. As expected, PD98059 (50 μM) inhibited p-ERK1/2 activation (Figure 4(A)) to a great extent (p < 0.01), Meanwhile, 30 μM SP600125 and 20 μM SB202190 were also used to display the inhibitory effects of JNK and p38 (p < 0.001, Figure 4(B) and (C)). Furthermore, the inhibitory effects of JNK and p38 were striking using 50 μM SP600125 and 40 μM SB202190, respectively.
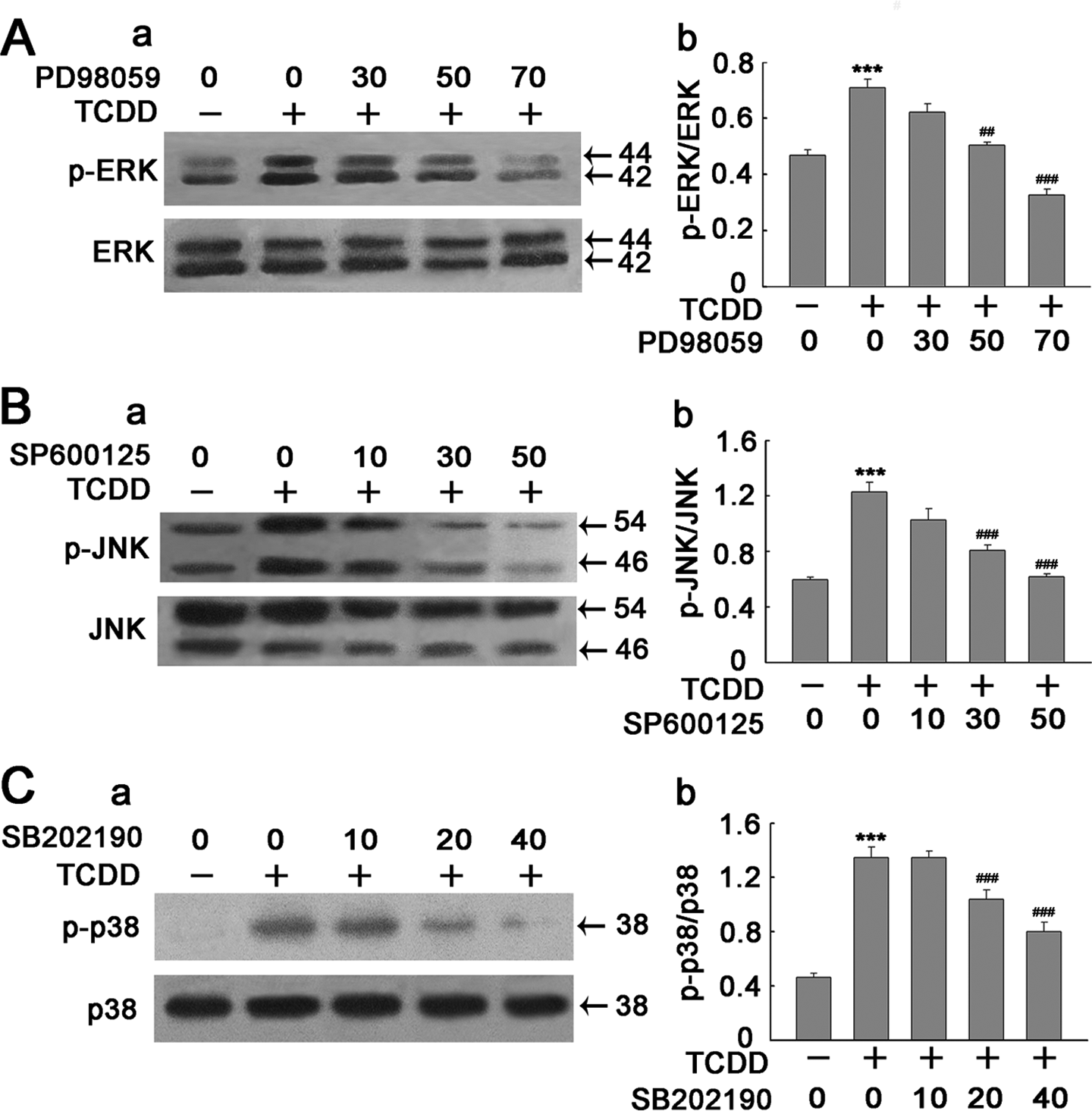
Inhibition of TCDD-induced phosphorylated-ERK1/2 (p-ERK1/2), p-JNK and p-p38 activation by inhibitors of MAPKs in PC12 cells. Cells were treated with TCDD in the absence or presence of PD98059 (30, 50 and 70 μM), SP600125 (10, 30 and 50 μM) and SB202190 (10, 20 and 40 μM). Cell extracts were subsequently prepared and subjected to Western blotting using antibody specific for p-ERK1/2, p-JNK, p-p38 and total-ERK (ERK1/2), JNK and p38. (A) (a), (B) (a) and (C) (a) The corresponding effects of MAPK inhibitors on TCDD-induced phosphorylation of ERK1/2, JNK and p38. (A) (b), (B) (b) and (C) (b) Denstiometric analysis for the results in (A) (a), (B) (a) and (C) (a), respectively. Data are presented as the mean ± SEM (n = 3). ***p < 0.001 compared to the control group. ## p < 0.01. ### p < 0.001 compared to the TCDD group. TCDD: 2,3,7,8-tetrachlorodibenzo-p-dioxin; MAPKs: mitogen-activated protein kinases; PC12: pheochromocytoma; ERK1/2: extracellular signal-regulated kinase1/2; JNK: c-Jun N-terminal kinase.
Specific inhibitive effects of MAPK inhibitors on TCDD-induced MAPKs phosphorylation
Specific inhibition of each signaling is important to consider the signal pathway. To ensure that MAPK inhibitors are specific for MAPKs in TCDD-treated PC12 cells, we examined the effects of these reagents on the phosphorylation of each target and other two molecules. As expected, 50 μM PD98059 profoundly inhibited ERK1/2 activation, while 30 μM SP600125 and 20 μM SB202190 had no effect on the ERK1/2 phosphorylation (Figure 5(A)). The similar results were found in the JNK and p38 activation, suggesting that each MAPK inhibitor (PD98059, SP600125 and SB202190) had no effect on one another.

Effects of each MAPK inhibitor on two other MAPKs. The cultures were pretreated with 50 μM PD98059, 30 μM SP600125 and 20 μM SB202190 for 1 h and followed by incubation with or without 200 nM TCDD for 3 h. Cell extracts were subsequently prepared and subjected to Western blotting using antibody specific for p-ERK1/2, p-JNK, p-p38 and total-ERK (ERK1/2), JNK and p38. (A) (a), (B) (a) and (C) (a) The corresponding effects of MAPK inhibitors on TCDD-induced phosphorylation of ERK1/2, JNK and p38. (A) (b), (B) (b) and (C) (b) Denstiometric analysis for the results in (A) (a), (B) (a) and (C) (a), respectively. Data are presented as the mean ± SEM (n = 3). ***p < 0.001 compared to the control group. ### p < 0.001 compared to the TCDD group. MAPK: mitogen-activated protein kinases; TCDD: 2,3,7,8-tetrachlorodibenzo-p-dioxin; p-ERK1/2: phosphorylated- extracellular signal-regulated kinase1/2; p-JNK: phosphorylated- c-Jun N-terminal kinase.
Inhibitory effects of MAPK inhibitors on apoptosis induced by TCDD
In addition, we detected the cell apoptosis in the MAPK inhibitor treated cells. According to the above results, the concentrations of 50 μM PD98059, 30 μM SP600125 and 20 μM SB202190 were used in subsequent experiments to examine the role of MAPKs activation in TCDD-treated PC12 cells. As shown in Figure 6(A), the three kinase inhibitors at micromolar concentrations individually caused a drastic increase in cell viability in TCDD-treated PC12 cells. FCM analysis demonstrated that apoptosis induced by TCDD was decreased by inhibitors of MAPKs (Figure 6(B)). Furthermore, the expression of active caspase-3 induced by TCDD was markedly attenuated by the kinase inhibitors (Figure 6(C)). Taken together, these results suggest that the three subgroups of MAPKs seem to play a positive role in the TCDD-mediated apoptosis in PC12 cells.

Effects of MAPK inhibitors on TCDD-induced apoptosis. The cultures were pretreated with 50 μM ERK1/2 inhibitor (PD98059), 30 μM JNK inhibitor (SP600125) and 20 μM p38 inhibitor (SB202190) for 1 h and followed by incubation with or without 200 nM TCDD for 24 h. (A) Cell viability was assessed using CCK-8 kit (n = 6). (B) PC12 cells were stained with Annexin-V and PI, followed by flow cytometry analysis. (C) (a) PC12 cells were pretreated MAPK inhibitors for 1 h and followed by incubation with TCDD or without TCDD for 12 h and then the cell lysates were analyzed by Western blotting using anti-caspase-3 antibodies (top) and anti-GAPDH antibodies (bottom). (b) Densitometer scan of active caspase-3/GAPDH ratio. ***p < 0.001 compared to the control group. ## p < 0.01. ### p < 0.001 compared to the TCDD group. MAPK: mitogen-activated protein kinases; TCDD: 2,3,7,8-tetrachlorodibenzo-p-dioxin; ERK1/2: extracellular signal-regulated kinase1/2; JNK: c-Jun N-terminal kinase; PC12: pheochromocytoma; CCK-8: Cell Counter Kit-8; GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
Discussion
TCDD is a prototypical environmental contaminant that can produce serious neurotoxicity in the nervous system. Our study demonstrated that in primary cortical neurons and PC12 cells, TCDD significantly caused apoptosis and suppressed cell viability in a dose- and time-dependent manner. Moreover, we found that TCDD could activate MAPK signaling pathways in TCDD-treated PC12 cells, and the effective attenuation of TCDD-induced apoptosis by the MAPK inhibitors provided additional support to the conclusion drawn in this study.
In this study, the cell viability assay performed by CCK-8 showed that TCDD effectively exerted cytotoxicity on primary cortical neurons and PC12 cells after 24 h treatment. The serious cell injuries caused by TCDD, such as nuclear fragmentation and even cell death, 25 may be accountable for the decrease in cell viability in primary cortical neurons, while the cell viability decrease in PC12 cells is possibly due to the cell growth inhibition and cell injuries caused by TCDD exposure. 26 Recently, it has been reported that TCDD can induce apoptosis by disruption of intracellular calcium homeostasis in human neuronal cell line SH-SY5Y. 27 After a 24-h culture, the characterization of TCDD toxicity in PC12 cells as an apoptotic process is supported by nuclear fragmentation, TUNEL assay in primary cortical neurons and FCM analysis in PC12 cells, which were both accompanied by the high level of active caspase-3. Caspase-3 is an executioner caspase, which leads to nucleosomal DNA fragmentation and plays a pivotal role in initiating apoptosis. 28
There are accumulating evidences indicating that TCDD can significantly induce apoptosis in various cell types. In the avian B-cells, TCDD causes AhR-mediated apoptosis, likely through activation of caspases-9 and executioner caspase-3.
29
TCDD can activate CYP450 oxidative enzymes, which lead to ROS production and subsequent apoptosis of chondrocytes.
30
On the other hand, studies performed using human lymphoblastic
MAPKs are important factors in the transduction of extracellular signals, from the membrane to the nucleus. 34 ERK1/2 mediates cell proliferation and differentiation and protects cells from apoptotic cell death, whereas JNK and p38, conversely, inhibit cell proliferation and may promote apoptotic cell death. 35 Our results showed that in PC12 cells, the activity of JNK and p38 underwent a quick upshift after TCDD treatment. The activated JNK has been reported to translocate to the nucleus where it can activate transcription factors in addition to c-Jun, such as p53 and c-Myc as well as non-transcription factors such as Bcl-2 and Bcl-xL. 36,37 The activated JNK also causes the release of apoptogenic factors, such as cytochrome c, and then directly regulates mitochondria-dependent apoptosis in a proapoptotic direction. 38,39 In addition, the activated p38 can activate downstream transcription factors, such as ATF-2 and the Max/Myc complex and stimulate the expression of active caspase-3 causing apoptosis. 40 In our study, we found that JNK inhibitor (SP600125) and p38 inhibitor (SB202190) notably suppressed cell apoptosis induced by TCDD treatment, and each MAPK inhibitor had no effect on one another. Moreover, we observed that the p38 inhibitor (SB202190) had a more significant inhibition compared with other two MAPK inhibitors, and it is indicated that the p38 pathway may be mainly involved in the TCDD-induced apoptosis of PC12 cells. These data suggest that JNK and p38 indeed play positive roles in the apoptosis induced by TCDD.
Our study showed that ERK1/2 was maximally phosphorylated at 6 h after TCDD treatment, while JNK and p38 were maximally phosphorylated at 3 h (Figure 3). However, it is unclear why cell death continued to increase during the later times, since ERK1/2 is considered to be protective to cell. 41 Moreover, we also found that when ERK1/2 inhibitor (PD98059) along with TCDD was used, apoptosis induced by TCDD was decreased instead of increased. So it was inferred that the role of ERK1/2 in this apoptotic process may be different from its general functions. It has been reported that ERK1/2 inhibitor (PD98059) pretreatment in Hepa-1c1c7 cells can reduce the expression of CYP1A1 gene induced by TCDD. 42 In PC12 cells, mitogen-extracellular kinase-1/2 (upstream of ERK1/2) inhibitor, U0126, decreases apoptosis of PC12 cells caused by cobalt chloride (CoCl2), which indicated that ERK1/2 may not act a protective role in CoCl2-induced injuries. 43 Additionally, ERK1/2 has been reported to potentiate apoptosis in leukemic cells. 44 So based on these studies, it can be concluded that ERK1/2 may act an unprotective role in this apoptotic process in TCDD-treated PC12 cells.
Although TCDD has been reported to activate MAPKs in many cell types, the mechanisms by which TCDD activates MAPKs still remain undefined. One of the most prominent signaling pathways to activate MAPKs is triggered by growth factor receptors. Indeed, TCDD has been reported to activate epidermal growth factor (EGF) receptor (EGFR)-associated tyrosine kinases in primary rat hepatocytes. 45,46 Similar activation of EGFR-mediated pathways can be activated by EGF or lipopolysaccharide, which cross-links the EGFR on the plasma membrane and initiate receptor-mediated signaling pathways, leading to MAPKs activation. 47,48 Therefore, TCDD may activate the MAPKs through signaling pathways similar to those employed by EGF. On the other hand, TCDD can trigger an immediate elevation of calcium influx rate and intracellular calcium mobilization in mouse hepatoma cells 49 and markedly increase intracellular calcium levels in neuroblastoma cells. 33 Since free intracellular Ca2+ is a key second messenger in cell signaling and has been directly linked to the induction of MAPKs, 50 it is inferred that MAPKs activation may be a result of TCDD-induced increase in intracellular Ca2+ levels. Moreover, studies in MCF10A human mammary epithelial cells further suggest a role of Ca2+ in TCDD-induced MAPKs activation. 51,52 Of course, there may be some other mechanisms to activate MAPKs by TCDD due to different cell types.
Most, if not all, of the biological effects of TCDD are mediated by the activation of AhR, which can produce many changes in gene transcription, such as CYP1A1 and CYP1B1 by translocating to the nucleus and binding to Ah-responsive elements (AHREs) in numerous genes. 53 As a transcription factor, AHREs typically located in the 5′-flanking region of target genes provides a platform for recruiting multiple coactivator proteins that enhance (or, in some cases, inhibit) gene transcription. 54 TCDD can induce the activation of MAPKs, which are believed to modulate transcription factor function and to regulate gene expression. However, the cross-talk between AhR and MAPK pathways in TCDD-induced toxicity is yet to be determined. Although the activation of ERK1/2 and JNK by TCDD does not require AhR, AhR is needed for the TCDD-induced upregulation of CYP1A1, indicating that AhR is a downstream rather than upstream of ERK1/2 and JNK. 55 Further support for these observations is a study using ERK1/2 inhibitor (PD98059) to reduce TCDD-induced AhR-DNA interactions. 42 However, in human keratinocytes, α-naphthoflavone (α-NF), an AhR antagonist, has been reported to attenuate TCDD-stimulated expression of JNK, which means that the JNK pathway is activated by TCDD in an AhR-dependent manner. 56 Moreover, in the absence of AhR, TCDD cannot induce p38 phosphorylation in rat and mouse hepatoma cells, demonstrating a pivotal role of AhR in the process of p38 activation by TCDD. 57 Consequently, it is assumed that AhR may play a distinctive role in the activation of three members of MAPKs and TCDD-induced cytotoxicity.
In conclusion, our experiments demonstrated that TCDD induced significant apoptosis in primary cortical neurons and PC12 cells. Besides, TCDD rapidly activated the MAPKs including ERK1/2, JNK and p38 in TCDD-treated PC12 cells. Furthermore, PD98059 (ERK1/2 inhibitor), SP600125 (JNK inhibitor) and SB202190 (p38 inhibitor) notably blocked the effect of TCDD on cell apoptosis and the induction of active caspase-3. Based on the above, it is concluded that MAPKs may play an important role in TCDD-induced neuronal apoptosis.
Footnotes
Authors’ Note
GX and ZD have contributed equally to this article.
Conflict of interest
The authors declared no conflicts of interest.
Acknowledgment
We greatly appreciate the technical support from Dr Tao Tao.
Funding
This work was supported by the National Natural Science Foundation of China (No.21077061 and No. 21277078).