
Abstract
Divalproex sodium is an antiepileptic drug. Hepatotoxicity is one of the most common side effects induced by divalproex sodium. Impaired fatty acid metabolism is considered to play an important role in the drug-induced hepatotoxicity. The sterol regulatory element-binding protein 1c (SREBP-1c) and peroxisome proliferator-activated receptor α (PPARα) are two key transcription factors involved, respectively, in fatty acid synthesis and degradation in liver. In the present study, we investigated the hepatotoxicity induced by divalproex sodium and its potential mechanism. The results indicated that divalproex sodium significantly decreased the cell viability and increased lactate dehydrogenase leakage in hepatocytes. The activities of alanine aminotransferase and aspartate transaminase were increased in hepatocytes treated with divalproex sodium. Furthermore, divalproex sodium activated SREBP-1c and increased the mRNA expressions of acetyl-CoA carboxylase 1, fatty acid synthase and stearoyl-CoA desaturase 1. Divalproex sodium also inhibited PPARα and decreased the messenger RNA expressions of 3-hydroxy-3-methylglutaryl-CoA synthase 2 and carnitine palmitoyltransferase 1A. These results suggest that the hepatotoxicity induced by divalproex sodium may be related with fatty acid synthesis and degradation mediated by SREBP-1c and PPARα in hepatocytes.
Introduction
Divalproex sodium is an antiepileptic drug. It is also used to treat mania and to help prevent migraine headaches. 1 Since its introduction in the market, divalproex sodium showed some side effects that have been reported in the clinic. 2,3 Hepatotoxicity is one of the most serious side effects induced by divalproex sodium, especially in children younger than 2 years old. 4 However, the mechanism of hepatotoxicity induced by divalproex sodium is not clear. So the present study focused on divalproex sodium–induced hepatotoxicity and its potential mechanism.
Impaired fatty acid metabolism is considered to play an important role in the drug-induced hepatotoxicity. 5 Divalproex sodium is composed of sodium valproate and valproic acid. 2 Our previous study has revealed that sodium valproate induced hepatotoxicity by elevating the gene expressions of cytochrome P450, family 1, subfamily A, polypeptide 1 (CYP1A1) and ATP binding cassette transporter G1 (ABCG1) related to fatty acid metabolism. 6 However, the effects of divalproex sodium on other gene expressions related to fatty acid metabolism in hepatocytes have not been elucidated yet. It has been reported that there are mainly two groups of transcription factors that mediate fatty acid metabolism in liver: the sterol regulatory element–binding proteins (SREBPs) and peroxisome proliferator–activated receptors (PPARs). 7 The mammalian genome encodes three SREBP isoforms, designated SREBP-1a, SREBP-1c and SREBP-2. The SREBP-1c participates in fatty acid synthesis by activating several enzymes including acetyl-CoA carboxylase 1 (ACC1), fatty acid synthase (FAS) and stearoyl-CoA desaturase 1 (SCD1). 8,9 SREBP-1c is involved in fatty acid synthesis, and the inhibition of PPARα function leads to impaired fatty acid degradation. PPARα has been proven to activate the genes including 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2) and carnitine palmitoyltransferase 1A (CPT1A), thereby improving fatty acid degradation. 10 Interestingly, the SREBP-1c is reported to interact and cross-talk with PPARα. 11,12 It has been reported that antipsychotic drugs including clozapine, olanzapine and haloperidol not only increased the expressions of SREBP-1c and its downstream genes ACC1, FAS and SCD1 but also decreased the expressions of PPARα and its downstream gene CPT1A in hepatocytes. 13,14 On the contrary, the study indicated that the hepatotoxicity induced by Fumonisin B1 was independent of PPARα. 15 Clofibrate causes an upregulation of PPARα target genes but does not alter the expression of SREBP target genes in the liver of pigs. 16
In the present study, the hepatotoxicity induced by divalproex sodium was assessed by measuring the cell viability, lactate dehydrogenase (LDH) leakage and the activities of alanine aminotransferase (ALT) and aspartate transaminase (AST) in hepatocytes. Then the messenger RNA (mRNA) expressions of SREBP-1c, ACC1, FAS, SCD1, PPARα, HMGCS2 and CPT1A were determined in hepatocytes treated with divalproex sodium.
Materials and methods
Materials
Dulbecco’s modified eagle’s medium (DMEM) and fetal bovine serum (FBS) were provided by Hyclone (Logan, Utah, USA). Collagenase B was purchased from Roche (Basel, Switzerland). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and dimethyl sulfoxide (DMSO) were purchased from Amersco (Solon, Ohio, USA). Other reagents were of the highest grade commercially available. All those primers were purchased from Applied Biosystems Company (California, USA).
Preparation of divalproex sodium
Divalproex sodium was synthetized by Tiansen Bioengineering and Technology Limited Company of Shaanxi in China. Divalproex sodium was prepared according to the procedures described below. 17,18
In 100 ml of acetone 16.6 g of sodium valproate and 14.4 g of valproic acid are dissolved at about 50°C. The solution is cooled to about 0°C for 5 h, filtered and the crystalline precipitate washed with 10 ml precooled acetone at about 0°C. Then 26.4 g white crystalline powder was obtained, which melts at 98.6–99.7°C. The new compound obtained showed a yield of 85.2%. Moreover, the infrared spectrum of the new compound is consistent with the standard infrared spectrum of divalproex sodium and has the following characterizing absorption bands: strong bands at 2960, 2873, 2935, 1694, 1569 and 1382 cm− 1. So the new compound was identified as divalproex sodium by its description, melting point and infrared spectrum.
Stock solutions of divalproex sodium were prepared in DMSO and freshly diluted to the desired concentrations before use. Final concentrations of DMSO in both control and treatment culture medium were identical in all studies, with a maximum level of 0.05%.
Primary cultures of rat hepatocytes
From male rats, the hepatocytes were isolated by two-step collagenase reperfusion (Sprague-Dawley, 240–280 g), as described previously. 19 Briefly, after anesthesia, a catheter was inserted into the hepatic portal vein and the liver was perfused with D-Hank’s solution to remove the blood. Then the liver was again perfused with D-Hank’s solution including collagenase B and calcium chloride. The liver was removed and minced in D-Hank’s solution, and the cellular suspension obtained was then purified by filtration, centrifugation and washed with D-Hank’s solution. Monolayer hepatocytes were cultured at 37°C in a 5% CO2 and 95% air atmosphere in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. Cell viability was consistently above 90%, as determined via Trypan blue exclusion.
Cell viability assay
The cell viability was assessed by a colorimetric MTT assay. 20 The hepatocytes were plated in a 96-well plate for 24 h. The different concentrations of divalproex sodium (100, 300, 1000 and 3000 μM) were added to each well and were cultured for 24 h. The cells were cultured for another 4 h after adding 20 μl MTT (5 mg/ml). Then the culture medium was removed and 150 μl of DMSO was added to dissolve the dye crystal. The absorbance change was evaluated spectrophotometrically at 490 nm using a Multiskan Ascent microplate reader (BMG, Offenburg, Germany). Cell viability was expressed as the percentage of the absorbance of treated cells relative to the absorbance of DMSO control cells.
LDH leakage assay
The release of LDH into the cell culture medium indicates cell membrane damage. 21 After incubation with different concentrations of divalproex sodium (100, 300, 500 and 1000 μM) for 24 h, the culture medium was collected for the estimation of LDH leakage using the LDH assay kit (Jiancheng Bioengineering Co. Ltd, Nanjing, China), according to the manufacturer’s protocols. LDH leakage was measured on the basis of oxidation of reduced form of nicotinamide adenine dinucleotide (NADH) to NAD+, and the change in absorbance was read at 440 nm in an ultraviolet/visible scanning spectrophotometer (TianMei UV-8500, Shanghai, China).
Assessment of ALT and AST activities
ALT and AST are two enzymes related with liver damage, as they are elevated in many liver diseases. The activities of ALT and AST were measured using a commercial kit by pyruvic acid method (Jiancheng Bioengineering Co. Ltd, Nanjing, China). After incubation with different concentrations of divalproex sodium (100, 300, 500 and 1000 μM) for 24 h, the culture medium was collected and the activities of ALT and AST were measured with automatic biochemistry analyzer (CL-7150, Hitachi, Tokyo).
RNA extraction and quantitative real-time polymerase chain reaction
After incubation with different concentrations of divalproex sodium (25, 50, 100, 200 and 500 μM) for 24 h, total cellular RNA was isolated from hepatocytes using TRIzol reagent (Invitrogen, California, USA). Complementary DNA (cDNA) was synthesized from 1 μg of total RNA using SuperScript III First Strand cDNA Synthesis Kit (Invitrogen, California, USA) according to the manufacturer’s protocol. The real-time polymerase chain reaction (PCR) was performed on an Applied Biosystems Company 7900 real-time PCR system (PE Biosystems, Carlsbad, USA), using SYBR Green PCR Master Mix (Invitrogen, California, USA). The fold changes in the levels of SREBP-1c, ACC1, FAS, SCD1, PPARα, HMGCS2 and CPT1A were corrected by the level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The 2−▵▵CT method was used to calculate the quantitation of relative gene expression as described by the manufacturer. The primers used for real-time PCR were listed in Table 1.
Primers used for quantitative real-time polymerase chain reaction (PCR)

ACC1: acetyl-CoA carboxylase 1; CPT1A: carnitine palmitoyltransferase 1A; FAS: fatty acid synthase; GADPH: glyceraldehyde-3-phosphate dehydrogenase; HMGCS2: 3-hydroxy-3-methylglutaryl-CoA synthase 2; PPARα: peroxisome proliferator-activated receptor α; SCD1: stearoyl-CoA desaturase 1; SREBP-1c: sterol regulatory element-binding protein 1c.
Data analysis
Data were expressed as mean ± standard deviation. Statistical analysis was carried out using one-way analysis of variance followed by Dunnett’s test. The values of p < 0.05 were considered statistically significant.
Results
Divalproex sodium decreased the viability of hepatocytes
To examine the effect of divalproex sodium on cell viability, the hepatocytes were treated with divalproex sodium for 24 h, the cell viability was assessed. As shown in Figure 1, divalproex sodium decreased the viability of hepatocytes in a dose-dependent manner. There were statistically significant differences in cell viability between the divalproex sodium groups (300 μM divalproex sodium group, p < 0.05; 1000 and 3000 μM divalproex sodium groups, p < 0.01) and the DMSO group.
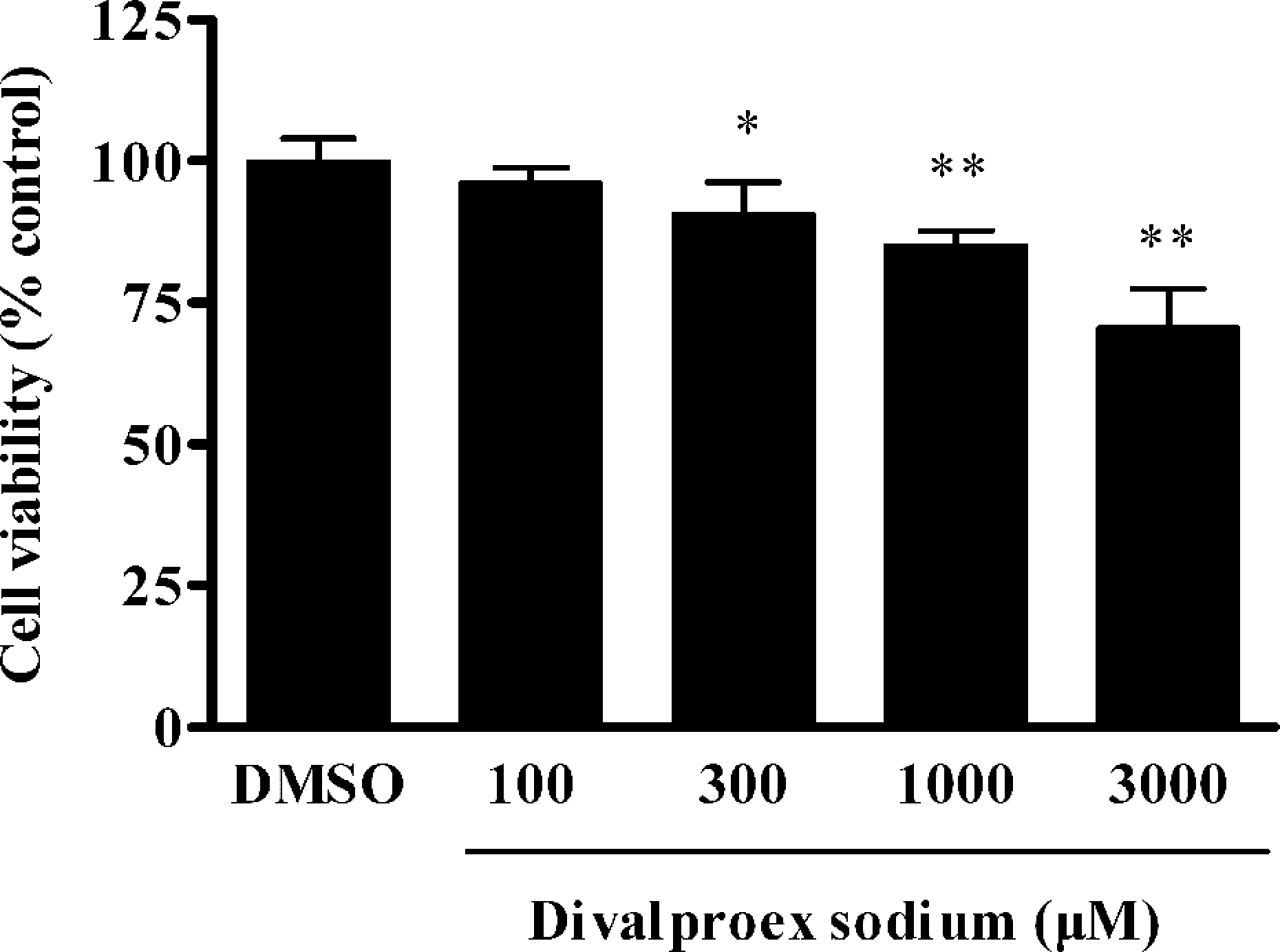
Divalproex sodium decreased the viability of hepatocytes. Hepatocytes were exposed to different concentrations of divalproex sodium (100, 300, 1000 and 3000 μM) for 24 h, cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Data were expressed as mean ± SD (n = 5). *p < 0.05 and **p < 0.01 compared with the dimethyl sulfoxide (DMSO) group.
Divalproex sodium increased LDH leakage
In order to investigate the effect of divalproex sodium on cell membrane integrity, the LDH level in the culture medium was measured. The results showed that divalproex sodium significantly increased LDH leakage in the culture medium of hepatocytes. There were statistically significant differences in LDH leakage between the divalproex sodium groups (100, 300, 500 and 1000 μM divalproex sodium groups, p < 0.01) and the DMSO group (Figure 2).

Divalproex sodium increased lactate dehydrogenase (LDH) leakage. The hepatocytes were exposed to different concentrations of divalproex sodium (100, 300, 500 and 1000 μM) for 24 h, the LDH leakage in culture medium was measured. Data were expressed as mean ± SD (n = 5). **p < 0.01 compared with the dimethyl sulfoxide (DMSO) group.
Divalproex sodium increased the activities of ALT and AST
In order to further investigate the effects of divalproex sodium on liver enzymes, the activities of ALT and AST in hepatocytes were measured. The hepatocytes were treated with different concentrations of divalproex sodium for 24 h and the activities of ALT and AST in culture medium were measured. As shown in Table 2, divalproex sodium increased significantly the activities of ALT and AST in hepatocytes in a dose-dependent manner. There were statistically significant differences in the activities of ALT and AST between the divalproex sodium groups (300 μM divalproex sodium group, p < 0.05; 500 and 1000 μM divalproex sodium groups, p < 0.01) and the DMSO group.
Divalproex sodium increased the activities of ALT and AST in hepatocytesa

ALT: alanine aminotransferase; AST: aspartate transaminase; DMSO: dimethyl sulfoxide.
aData were expressed as mean ± SD (n= 5).
b p < 0.05 compared with the DMSO group.
c p < 0.01 compared with the DMSO group.
Divalproex sodium increased the mRNA expressions of SREBP-1c and its downstream genes
To evaluate whether the hepatotoxicity induced by divalproex sodium was to activate the transcription factor SREBP-1c and its downstream genes, we measured the mRNA expressions of SREBP-1c, ACC1, FAS and SCD1 by real-time PCR. The results showed that the SREBP-1c mRNA expression was significantly increased in a dose-dependent manner (Figure 3(a)). There were statistically significant differences in the mRNA expression of SREBP-1c between the divalproex sodium groups (100 μM divalproex sodium group, p < 0.05; 200 and 500 μM divalproex sodium groups, p < 0.01) and the DMSO group. Moreover, these data also showed that divalproex sodium increased the mRNA expressions of ACC1, FAS and SCD1 about 2 times in 500 μM divalproex sodium group as compared to the DMSO group (p < 0.01; Figure 3(b), (c) and (d)).

Divalproex sodium increased the expressions of sterol regulatory element-binding protein 1c (SREBP-1c) and its downstream genes acetyl-CoA carboxylase 1 (ACC1), fatty acid synthase (FAS) and stearoyl-CoA desaturase 1 (SCD1) in hepatocytes. The hepatocytes were exposed to different concentrations of divalproex sodium (25, 50, 100, 200 and 500 μM) for 24 h. The messenger RNA (mRNA) expressions were measured by real-time polymerase chain reaction (PCR). (a) Effect of divalproex sodium on SREBP-1c expression. (b) Effect of divalproex sodium on ACC1 expression. (c) Effect of divalproex sodium on FAS expression. (d) Effect of divalproex sodium on SCD1 expression. Data were expressed as mean ± SD (n = 3). *p < 0.05 and **p < 0.01 compared with the dimethyl sulfoxide (DMSO) group.
Divalproex sodium decreased the mRNA expressions of PPARα and its downstream genes
To further evaluate whether the hepatotoxicity induced by divalproex sodium was related with the transcription factor PPARα and its downstream genes, we measured the mRNA expressions of PPARα, HMGCS2 and CPT1A by real-time PCR. As a result, divalproex sodium decreased the mRNA expression of PPARα in a dose-dependent manner (Figure 4(a)). There were statistically significant differences in the mRNA expression of PPARα between the divalproex sodium groups (50 μM divalproex sodium group, p < 0.05; 100, 200 and 500 μM divalproex sodium groups, p < 0.01) and the DMSO group. Moreover, divalproex sodium decreased the mRNA expressions of HMGCS2 and CPT1A. There were statistically significant differences in the expressions of HMGCS2 and CPT1A between the divalproex sodium groups (100 μM divalproex sodium group, p < 0.05; 200 and 500 μM divalproex sodium groups, p < 0.01) and the DMSO group (Figure 4(b) and (c)).

Divalproex sodium decreased the expressions of peroxisome proliferator-activated receptor α (PPARα) and its downstream genes 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2) and carnitine palmitoyltransferase 1A (CPT1A) in hepatocytes. The hepatocytes were exposed to different concentrations of divalproex sodium (25, 50, 100, 200 and 500 μM) for 24 h. The messenger RNA (mRNA) expressions were measured by real-time polymerase chain reaction (PCR). (a) Effect of divalproex sodium on PPARα expression. (b) Effect of divalproex sodium on HMGCS2 expression. (c) Effect of divalproex sodium on CPT1A expression. Data were expressed as mean ± SD (n = 3). *p < 0.05 and **p < 0.01 compared with the dimethyl sulfoxide (DMSO) group.
Discussion
Drug-induced hepatotoxicity is a major problem during drug development and the main cause for drug withdrawals from the market. 22 Therefore, it is necessary to evaluate drug-induced hepatotoxicity for the design of safer therapeutic agents. Clinical studies have shown that divalproex sodium caused hepatic failure in patients accompanied by the abnormalities of hematology, serum chemistry and routine urinalysis. Even some patients progressed to fatal hepatotoxicity without developing any specific hepatic function abnormalities. 4 Although there have been numerous investigations into the mechanisms of hepatotoxicity of sodium valproate and valproic acid, the exact mechanism of hepatotoxicity induced by divalproex sodium was not clear. So it is very significant to explore divalproex sodium-induced hepatotoxicity and its potential mechanism. The cell viability, a marker of cytotoxicity, was decreased in divalproex sodium-treated hepatocytes in a dose-dependent manner in our present experiments. The LDH leakage is also a biomarker of cell integrity. LDH catalyzes the reversible oxidation of L-lactate to pyruvate; the increase in LDH leakage in hepatocytes confirms increased hepatocyte membrane permeability and cellular leakage. 23 Clinical study confirmed that the LDH level in plasma was increased in patients treated with divalproex sodium. 23,24 Our results showed that divalproex sodium increased LDH leakage in hepatocytes. In previous experiments, we have observed that sodium valproate decreased the cell viability and increased LDH leakage in human hepatocellular liver carcinoma (HepG2) cells. 6 In addition, the increase in ALT and AST activities can be interpreted as evidence of hepatotoxicity. In the present study, we found that divalproex sodium significantly increased the activities of ALT and AST in hepatocytes in a dose-dependent manner. Clinical studies have also reported that divalproex sodium increased the activities of ALT and AST in patients. 25,26 In a previous study, we have observed sodium valproate increased the activities of ALT and AST in HepG2 cells. 6 Taken together, accumulating data suggest that divalproex sodium can result in the damage of hepatocytes and finally cause cell death.
It has been demonstrated that the transcription factors SREBPs and PPARs participate in fatty acid synthesis and degradation in liver, respectively. 27 SREBP-1c is by far the most abundant isoform in liver and mainly regulates the expression of genes involved in fatty acid synthesis. 28,29 In the present study, we found that divalproex sodium increased the expression of SREBP-1c in hepatocytes. As well, the study showed that the antipsychotic drugs clozapine and haloperidol stimulated fatty acid synthesis through the activation of SREBP-1c. 30 The enzymes of ACC1, FAS and SCD1 have been confirmed to participate in fatty acid synthesis regulated by SREBP-1c in liver. ACC1 is a biotin-containing enzyme which catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, the rate-limiting step in fatty acid synthesis. 31 The FAS plays an important role in lipogenesis of mammals through the synthesis of saturated long-chain fatty acids from acetyl-CoA and malonyl-CoA. 32,33 SCD1 is an enzyme responsible for the conversion of saturated fatty acid into monounsaturated fatty acids. 34 Our present results demonstrated that divalproex sodium increased the mRNA expressions of ACC1, FAS and SCD1 in hepatocytes. It has been observed that the antipsychotic drugs clozapine and olanzapine strongly increased fatty acid synthesis by activating SREBP-1c and its associated downstream genes ACC1, FAS and SCD1 in rat hepatocytes. 14 The experiments also showed that the antipsychotics and antidepressants including haloperidol, olanzapine and mirtazapine induced hepatotoxicity by activating SREBP-1 and downstream genes HMGCS1 and SCD1 in cultured human liver cells. 35 In addition, ethanol induced the hepatotoxicity by activating SREBP-1c and its downstream genes ACC and SCD1 involved in the synthesis of fatty acid in rats. 36 Data from the present study suggest that the hepatotoxicity induced by divalproex sodium may be related with fatty acid synthesis mediated by SREBP-1c and its downstream genes ACC1, FAS and SCD1 in hepatocytes.
In addition to fatty acid synthesis mediated by SREBP-1c, the fatty acid degradation mediated by PPARα also plays a major role in drug-induced hepatotoxicity. 37 In the present study, our data indicated that divalproex sodium decreased the expression of PPARα in hepatocytes. The antipsychotic drug including chlorpromazine, clozapine and ziprasidone were reported to decrease the fatty acid degradation by inhibiting PPARα in mice. 38 Furthermore, PPARα regulates fatty acid degradation by activating its downstream genes HMGCS2 and CPT1A in liver. In fact, the activation of both HMGCS2 and CPT1A is a means of removing excess hepatic fatty acid. 39,40 The HMGCS2 gene is the most responsive to PPARα among its downstream genes. It is the first and rate-limiting enzyme in ketogenesis of liver. 41 We further found that divalproex sodium decreased the expression of HMGCS2 in hepatocytes. And that, divalproex sodium decreased the expression of CPT1A in hepatocytes. CPT1A is the most abundant enzyme in liver. It catalyzes the esterification of acylcoenzyme A to carnitine, which is a key step in fatty acid oxidation. 42,43 As a matter of fact, the CPT1A is upregulated by PPARα in human liver. 44 The antipsychotic drugs clozapine decreased the expressions of PPARα and its downstream gene acyl-CoA oxidase 1 in rat liver. 13 Ethanol has been reported to cause acute liver damage by decreasing PPARα and its downstream genes medium chain acyl-CoA dehydrogenase and acyl-CoA oxidase expressions of liver tissues in mice. 45 Euchresta horsfieldii Benn., which has been used for the traditional treatment of hyperlipidemia, increased the expressions of PPARα and its downstream genes HMGCS2 and CPT1A in human HepG2 cells. 46 Overall, these findings indicate that the hepatotoxicity induced by divalproex sodium may be related with fatty acid degradation mediated by PPARα and its downstream genes HMGCS2 and CPT1A in hepatocytes.
In summary, the present study suggests that divalproex sodium can induce hepatotoxicity by decreasing cell viability and elevating the liver enzymes such as LDH, ALT and AST. Moreover, the hepatotoxicity induced by divalproex sodium may be related with both fatty acid synthesis mediated by SREBP-1c and fatty acid degradation mediated by PPARα. Studies are needed to measure the changes in these relevant proteins. Further studies are necessary to identify the exact mechanism of hepatoxicity induced by divalproex sodium.
Footnotes
Funding
This study was supported by the National Natural Science Foundation of China (No. 81072643), Scientific and technological project of Xi’an city, Shaanxi Province (No. SF1028 (1)) and Doctoral Fund of Ministry of Education of China (No. 20090201110050).
Declaration of Conflict of Interest
The authors declared no conflicts of interest.