
Abstract
Background:
Patients on peritoneal dialysis (PD) may suffer from sodium (Na) and fluid overload, hypertension and increased cardiovascular risk. Low-Na dialysis solution, by increasing the diffusive removal of Na, might improve blood pressure (BP) management.
Methods:
A glucose-compensated, low-Na PD solution (112 mmol/L Na and 2% glucose) was compared to a standard-Na solution (133 mmol/L Na and 1.5% glucose) in a prospective, randomised, single-blind study in hypertensive patients on PD. One daily exchange of the standard dialysis regimen was substituted by either of the study solutions for 6 months. The primary outcome (response) was defined as either a decrease of 24-h systolic BP (SBP) by ≥6 mmHg or a fall in BP requiring a medical intervention (e.g. a reduction of antihypertensive medication) at 8 weeks.
Results:
One hundred twenty-three patients were assessed for efficacy. Response criteria were achieved in 34.5% and 29.1% of patients using low- and standard-Na solutions, respectively (p = 0.51). Small reductions in 24 h, office, and self-measured BP were observed, more marked with low-Na than with standard-Na solution, but only the between-group difference for self-measured SBP and diastolic BP was significant (p = 0.002 and p = 0.003). Total body water decreased in the low-Na group and increased in the control group, but between-group differences were not significant. Hypotension and dizziness occurred in 27.0% and in 11.1% of patients in the low-Na group and in 16.9% and 4.6% in the control group, respectively.
Conclusions:
Superiority of low-Na PD solution over standard-Na solution for control of BP could not be shown. The once daily use of a low-Na PD solution was associated with more hypotensive episodes, suggesting the need to reassess the overall concept of how Na-reduced solutions might be incorporated within the treatment schedule.
Keywords
Introduction
The prevalence of arterial hypertension among patients with chronic kidney disease undergoing long-term dialysis treatment has been estimated at around 80%. 1 It is among the leading risk factors of atherosclerotic cardiovascular morbidity and mortality in dialysis. 2,3
In dialysis patients, sodium (Na) and water overload are the most important contributing factors to the pathogenesis of hypertension. 4 The impact of peritoneal Na elimination during peritoneal dialysis (PD) on blood pressure (BP) has been comprehensively described, 5 and the restoration of the Na balance has long been recognised as a primary aim of dialysis treatment. 6 However, although water and Na are removed continuously during PD, patients nevertheless remain in a state of overhydration related to net Na imbalance. 7,8
By lowering the Na concentration of the dialysis fluid, 9 –12 Na elimination can be improved through diffusive transport from blood to the peritoneal cavity. A previous clinical study investigated the most adequate composition (Na/glucose) in a 2-month treatment with Na concentrations of 115 and 102 mmol/L, with and without the adjustment of glucose concentration. Only the Na concentration of 115 mmol/L with glucose compensation was effective on BP, primarily through decrease of night-time BP. Low-Na dialysis solutions require glucose augmentation to maintain an osmolarity that prevents a loss of ultrafiltration, which could offset the gain in Na elimination achieved through diffusion. 13
We performed a randomised, prospectively controlled trial to investigate the effect of a once-daily dwell with a glucose-compensated low-Na solution in hypertensive PD patients. The objective was to demonstrate superiority of the low-Na solution over a standard solution regarding the lowering of BP.
Methods
Study design
We report on a prospective, single-blind, controlled, randomised, parallel-group, multicentre phase III study in hypertensive patients suffering from chronic renal failure and being treated with continuous ambulatory PD (CAPD) or automated PD (APD), with or without antihypertensive drugs, including diuretics. The study compared the effect on BP of an investigational low-Na PD solution to that of a standard-Na solution. The procedures started with a 4- to 8-week preparation phase during which the patients’ eligibility for participation was confirmed. They were stabilised on their currently prescribed standard dialysis treatment regimen using the reference standard-Na solution and on BP medication. At the end of the preparation phase, eligible patients were randomised and entered a 6-month single-blind treatment phase during which the standard glucose bag of a single daytime dwell was replaced by either the low-Na solution or the standard-Na solution as control. The assessment of efficacy was based on the first 8 weeks of randomised treatment (‘efficacy period’) following regulatory guidance for efficacy assessment of antihypertensive drugs. 14 The entire exposure period of 6 months served as safety period. Individual study participation ended with a 2-month follow-up phase on standard PD prescription (Supplemental Figure 1). Assessments were performed monthly until the end of follow-up.
Participants and randomisation
Eligible patients were ≥18 years old and suffered from chronic kidney disease treated with CAPD or APD for at least 3 months. Participants on CAPD had to have ≥3 bag exchanges per day, those on APD at least one daytime exchange (with the rest of the day dry or wet). For both modalities, at least one exchange of 1.5% glucose with a dwell time of 4 6 h was required to correspond to the investigational product at low glucose strength. Moreover, patients either had to present with an office systolic blood pressure (SBP) ≥140 mmHg or with a diastolic blood pressure (DBP) ≥90 mmHg or had to be stabilised on antihypertensive medication (including diuretics). Patients with low BP (office SBP < 120 mmHg, confirmed by an average 24-h SBP ≤105 mmHg determined by ambulatory blood pressure measurement (ABPM)) as well as those with orthostatic hypotension (i.e. fall in office SBP of ≥20 mmHg after standing for ≥1 min) or with hyponatremia <130 mmol/L were excluded. Patients were also ineligible if they suffered from chronic arrhythmia, had had peritonitis within 1 month before enrolment, or had a life expectancy <9 months. Patients were allocated to the investigational treatments at a ratio of 1:1 using a concealed, centralised block randomisation stratified by country and mean 24-h SBP >130 mmHg versus ≤130 mmHg, as determined by ABPM.
Interventions
The composition of the investigational products is given in Supplemental Table 1. The products were available in three compartment bags of which only compartments A and C were to be used (the inadvertent use of compartment B was prevented by means of an unbreakable pin). After mixing of the compartments, the composition of both products was identical except for Na and glucose with 112 mmol/L Na and 2.0% glucose in the low-Na and 133 mmol/L Na and 1.5% glucose in the standard-Na solution, respectively.
During randomised treatment, one daytime dwell of the standard PD prescription (preferably the last dwell during the day, with a dwell time of 4 ± 1 h) was replaced by the investigational product. For patients on APD mode, this was applied by a manual CAPD exchange before the nightly treatment. Adjustments in the standard PD treatment were allowed if considered necessary by the investigator during the run-in and again after month 2 of randomised treatment. Change of antihypertensive medication was not permitted during the efficacy period, except in case of hypotension (response criterion, see ‘Outcomes’ section).
Outcomes
The primary outcome measure for efficacy was the proportion of treatment responders. Response was defined as either (a) a decrease of the mean 24-h SBP from baseline to week 8 by at least 6 mmHg, a difference shown to be relevant for reducing cardiovascular events, 3 without modifications in antihypertensive medication or (b) a fall in BP requiring a medical intervention, such as a decrease of antihypertensive medication, and confirmed by the study’s Data Safety Monitoring Board. The board periodically reviewed the data for BP, antihypertensive medication, adverse events (AEs), and PD prescriptions based on blinded data.
Secondary outcomes for BP included 24-h ABPM of daytime and night-time SBP and DBP
ABPM was performed twice using a Spacelabs 24-h ABPM 90207/90217 device (validated by the British Hypertension Society 15 ) with readings at 15-min intervals between 6 a.m. and 10 p.m. and at 30-min intervals, otherwise, that is, once before randomisation as well as once after 8 weeks (within a window between 7 weeks and 9 weeks after start of randomised treatment). Office BP measurements (mean of at least two measurements in sitting position after at least 5 min of rest with Omron HEM 907 device) and a check for orthostatic hypotension were performed at each study visit. Moreover, the subjects had to perform self-measurements of BP using an Omron M10-IT device on the same arm and site used for measuring office BP. Self-measurements were averaged from six daily measurements recorded in the patient diary performed in the morning (before breakfast, intake of antihypertensive medication and first PD bag exchange) and evening (close to bedtime, before intake of antihypertensive medication, and as far as possible after the last PD bag exchange) in a sitting position on the 3 days preceding each visit. Single-dwell and estimated 24-h Na removal, total body water (TBW), extracellular water (ECW) and intracellular water (ICW) were determined by whole body bioimpedance spectroscopy (Body Composition Monitor, Fresenius Medical Care, Bad Homberg, Germany) and by measuring body weight.
Safety outcomes were spontaneously reported AEs, solicited AEs of special interest (hyponatremia, hypotension, dizziness, asthenia, peritonitis and any events leading to changes in treatment during the study), safety laboratory measures and vital signs, as well as residual renal function.
Statistical methods and sample size
The primary analysis of efficacy was based on the full analysis set (FAS), which included all randomised patients who attended the baseline visit. As prespecified in the statistical analysis plan, a Cochran–Mantel–Haenszel test stratified for mean screening 24-h SBP (≤130 mmHg vs. >130 mmHg) was computed for comparing the treatment groups’ response rates using a two-sided type I error level of α = 0.05. A 95% confidence interval for the difference in response rates between treatment groups was provided additionally using the Newcombe–Wilson score method. For the confirmatory analysis of the primary outcome measure, no missing data imputation was performed, that is, the primary analysis was based on patients with valid BP response data. Sensitivity analyses were performed in the FAS, in which all patients with missing data were considered either responders or non-responders as well as in the per-protocol analysis data set (PPS), which included all FAS-eligible patients who completed the first 2 months of randomised treatment without major protocol deviations or who had a medically important fall in BP during the first 2 months.
Secondary efficacy and safety outcomes were analysed using applicable methods of descriptive data analysis depending upon their level of measurement. For metric secondary efficacy outcomes, descriptive analysis of covariance (ANCOVA) models were computed using the baseline value of the dependent variable as a covariate and treatment, mean 24-h SBP (≤130 mmHg vs. >130 mmHg), and country as factors. For secondary outcomes, p-values ≤0.05 were considered descriptively significant without multiplicity adjustment.
The sample size estimation was based on expected response rates of 65% and 40% for low-Na and standard-Na solution, respectively. Using a χ 2 -test model, a type I error level of α = 0.05 (two-sided) and 1:1 randomisation, a total of at least 125 efficacy evaluable subjects were required to assure a power of 80% for demonstrating superiority of the low-Na solution.
Results
Recruitment and participant flow
Between August 2008 and December 2014, a total of 158 patients were enrolled and 128 were randomised (low-Na 63 and standard-Na 65). All randomised patients were evaluated for safety (safety analysis set). Five randomised patients were withdrawn from the trial due to an AE before the baseline visit and were thus removed from the FAS, which included 60 patients assigned to low-Na and 63 patients assigned to standard-Na. A total of 88 patients (low-Na 45 and standard-Na 43) completed the randomised part of the study as scheduled (Supplemental Figure 2).
Thirty-six patients in each treatment group qualified for the PPS. The most frequent protocol deviations leading to exclusion from the PPS (multiple responses) were conduct of the visit scheduled at week 8 outside the acceptable window (low-Na 9 and standard-Na 12), missing response assessment (7 and 12 patients) and discrepancies between the date of ABPM assessment and the applicable visit date (10 and 5 patients).
Participant characteristics and PD history
The characteristics of the patients in the FAS are provided in Table 1. Despite some slight differences between both treatment groups at baseline, demographic and anthropometric characteristics were essentially comparable as well as measures that might impact efficacy like renal function. For creatinine clearance, the baseline mean value difference was attributable to individual outlying values while the groups’ medians were comparable.
Patient characteristics at baseline (full analysis set).
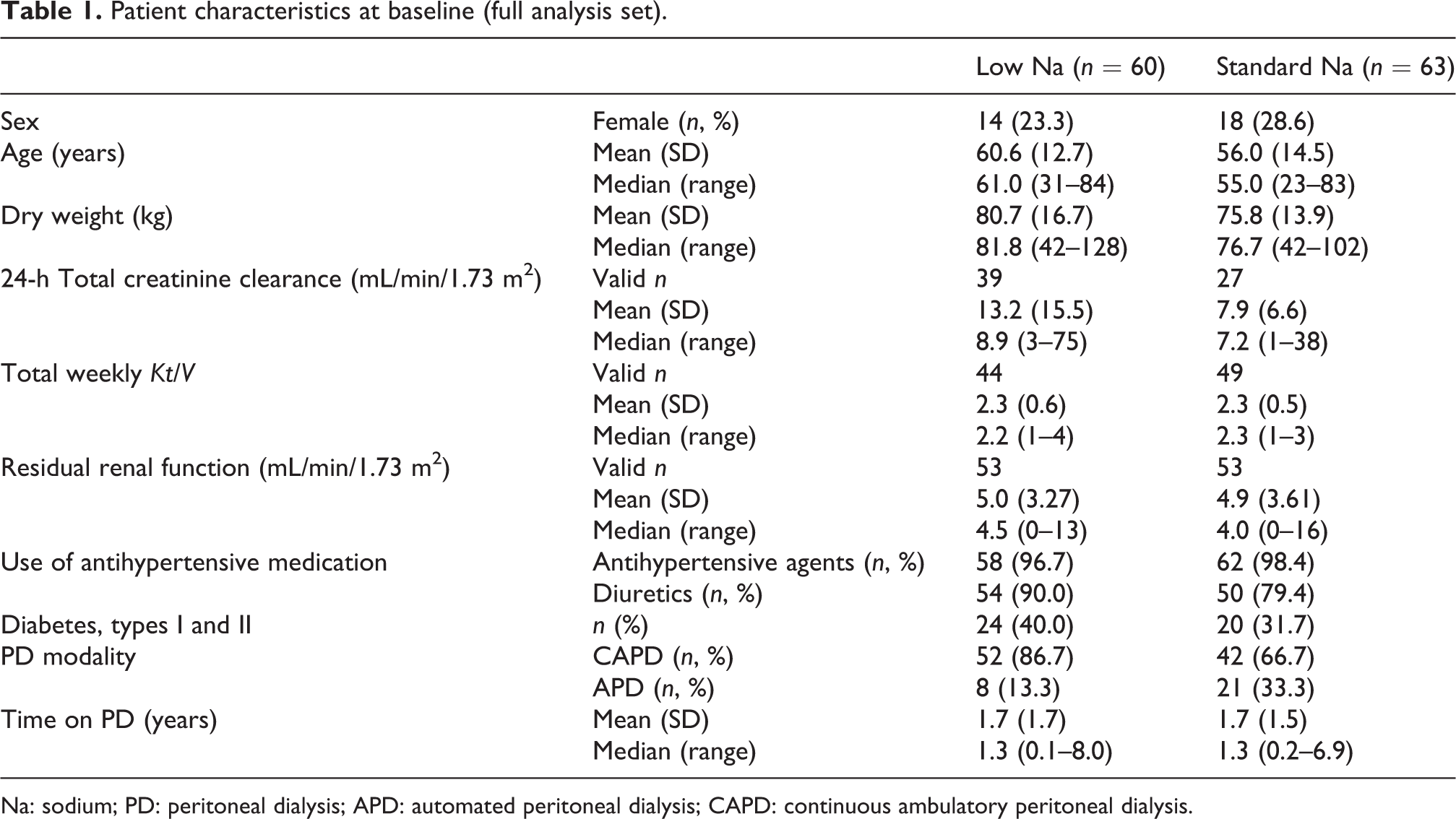
Na: sodium; PD: peritoneal dialysis; APD: automated peritoneal dialysis; CAPD: continuous ambulatory peritoneal dialysis.
There was a slight imbalance between the treatment groups regarding PD modality (see Table 1), but the treatment groups were essentially comparable with regard to all relevant parameters of the PD prescription (mean glucose concentration, dwell time and number of exchanges).
Efficacy
Improvement of hypertension (primary outcome measure)
The number and percentage of patients who met the response criteria for improvement of hypertension are provided in Table 2. Superiority of the new formulation over standard-Na solution could not be confirmed statistically (p = 0.512). Our sensitivity analysis in which patients with missing response data were counted as either responders or non-responders, as well as the analysis in the PPS (p = 0.296), led to the same conclusion.
Improvement of hypertension – responders (full analysis set; number and % of patients with valid data, rate difference and 95% CI, Cochran–Mantel–Haenszel test p-value stratified by screening 24-h systolic blood pressure ≤130 mmHg vs. >130 mmHg).
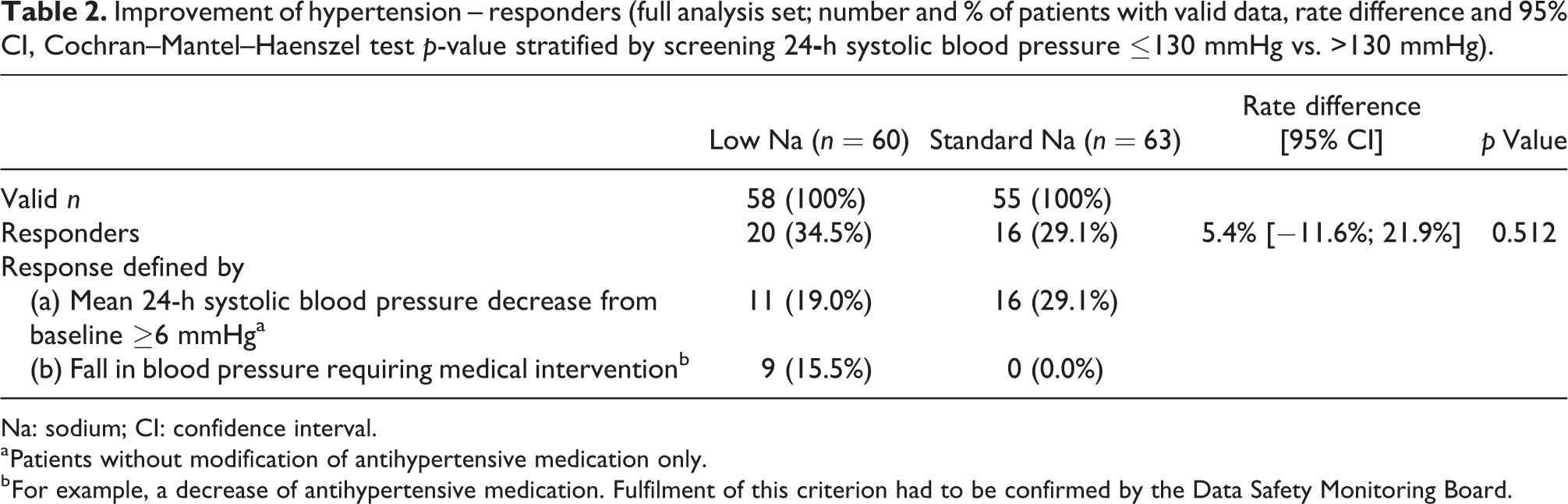
Na: sodium; CI: confidence interval.
a Patients without modification of antihypertensive medication only.
b For example, a decrease of antihypertensive medication. Fulfilment of this criterion had to be confirmed by the Data Safety Monitoring Board.
Table 2 also provides that a fall in BP necessitating a medical intervention such as a reduction of antihypertensive medication was observed only in patients receiving low-Na solution, whereas fewer patients on low-Na were counted for the ≥6 mmHg SBP decrease criterion as compared to standard-Na.
In a subgroup analysis, patients with screening 24-h SBP ≤130 mmHg in 7 of 21 evaluable subjects (33.3%) in the low-Na group were responders (six of them requiring a reduction of antihypertensive medication), as compared to 1 of 19 patients in the standard-Na group (p = 0.03). No appreciable treatment group difference in response rates was observed in patients with initial 24-h SBP >130 mmHg (p = 0.57). Further post hoc subgroup analysis performed to assess possible influences of baseline treatment group differences regarding PD modality and the percentage of patients with diabetes (see Table 1) on the primary outcome measure revealed no indication of bias (data not shown).
Serial BP measurements and antihypertensive medication
During the 8-week efficacy period, both treatment groups showed non-significant decreases of SBP and DBP according to 24-h ABPM and office measurements as well as the patients’ self-measurements showed significant decreases of SBP and DBP for low-Na (p = 0.004 and p = 0.008) but increases for standard-Na (Table 3 and Figure 1), with significant differences between the treatment groups for both SBP and DBP (p = 0.002 and p = 0.003). According to 24-h ABPM, the SBP and DBP decreases in the low-Na group were more pronounced during night-time, following the application of the study bag as last bag of the day than during day-time while the opposite was the case in the standard-Na group.
Secondary efficacy outcomes – baseline/screening value and absolute intraindividual change (full analysis set; mean (standard deviation) and patients with valid values).
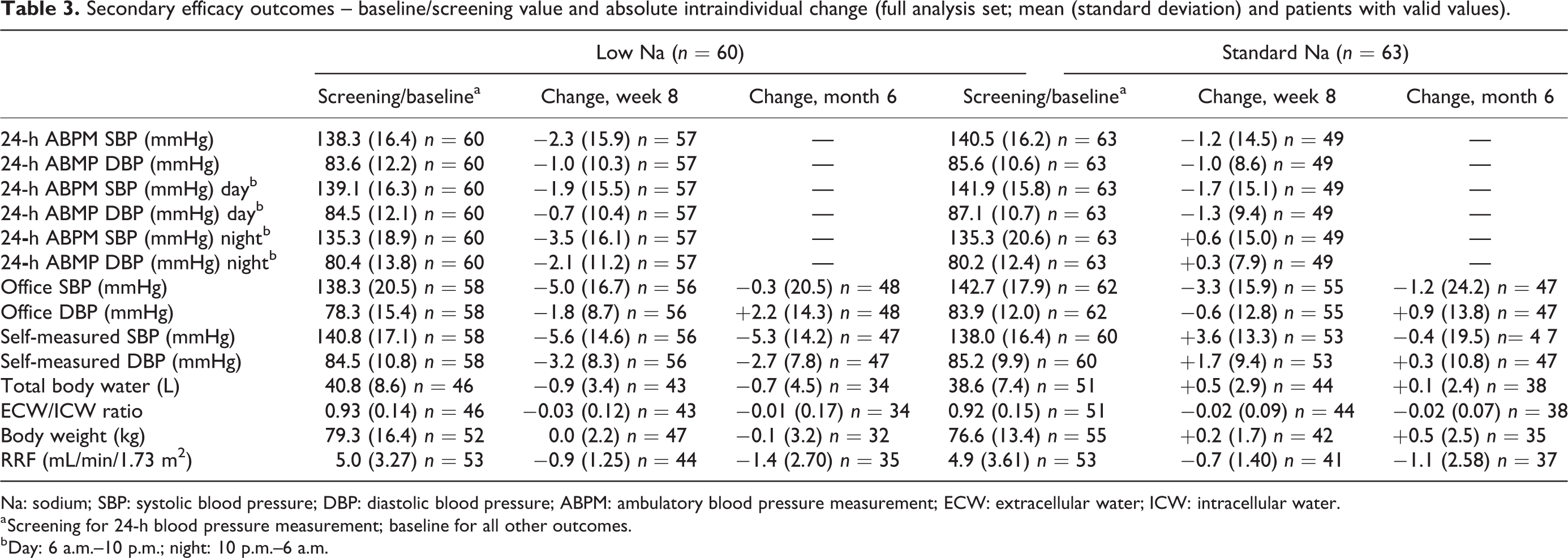
Na: sodium; SBP: systolic blood pressure; DBP: diastolic blood pressure; ABPM: ambulatory blood pressure measurement; ECW: extracellular water; ICW: intracellular water.
a Screening for 24-h blood pressure measurement; baseline for all other outcomes.
b Day: 6 a.m.–10 p.m.; night: 10 p.m.–6 a.m.
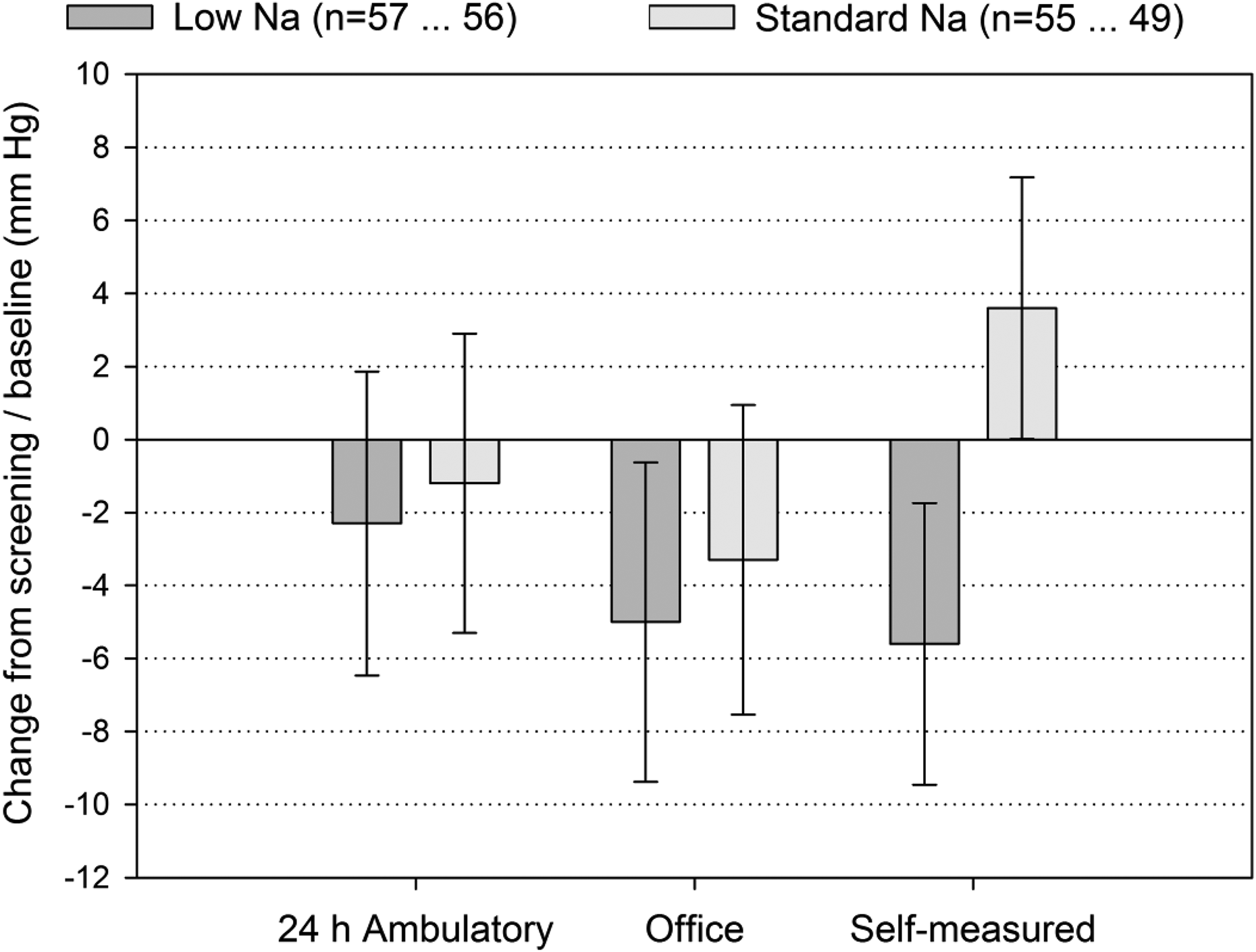
Systolic blood pressure change between screening (24 h) or baseline (office, self-measured) and week 8 (full analysis set; means and 95% confidence intervals).
While the office BP measurements suggest that the reductions observed during the efficacy period abated during the remainder of randomised treatment, the patients’ self-measurements of BP indicate that the decreases in the low-Na group were preserved until at least month 6 after which the patients were switched back to their previous prescription (Figure 2).
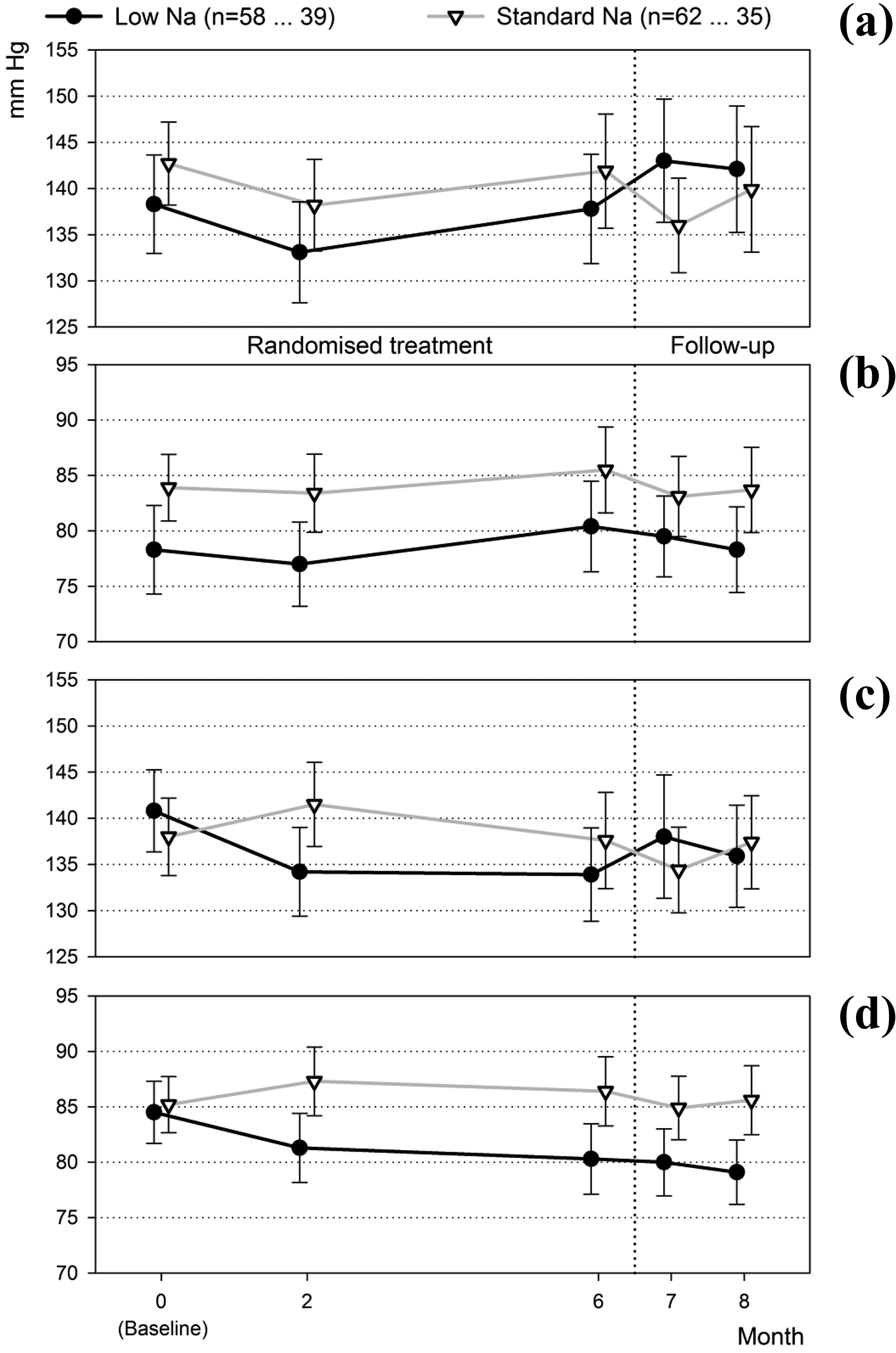
Blood pressure time course – office (a) systolic, (b) diastolic, (c) self-measured systolic and (d) self-measured diastolic (full analysis set; means and 95% confidence intervals).
Decreases in antihypertensive medication were reported for seven patients (11.7%) in the low-Na group during the first 4 weeks of randomised treatment and again for seven patients (11.7%), some of whom were the same individuals between weeks 5 and 8, compared to 2 (3.2%) and 1 (1.6%) patient for standard-Na. During the same periods increases were documented in one (1.7%) and two (3.3%) patients in the low-Na group and in two (3.2%) and six (9.5%) patients for standard-Na. During months 3 through 6, the average change of number of antihypertensive drugs (including diuretics) per patient was from 3.3 (1.5) (mean, SD) to 3.0 (1.4) for low-Na and from 3.3 (1.4) to 3.4 (1.5) for standard-Na.
Na removal, hydration and body weight
Data for Na removal were available for fewer than 15 patients in each group and were thus considered to be not representative for the study population. TBW decreased slightly in the low-Na group but not in the standard-Na group, whereas slight but statistically significant decreases of the ECW/ICW ratio were observed in both groups between baseline and week 8 (ANCOVA models: p < 0.05; Table 3). Treatment group differences for TBW and ECW/ICW ratio were not significant. Both groups showed no relevant changes in body weight.
Safety/tolerability
Table 4 provides an overview of AEs with onset between the beginning and end of randomised treatment. Even though the overall numbers of events and of patients with events were comparable between the study groups, a larger percentage of patients had potentially treatment-related events and special interest events in the low-Na group. One event in each group was fatal, assessed as unlikely or not related to the standard-Na and the low-Na solution, respectively. Subjects experiencing serious AEs were mostly affected by infections (peritonitis), gastrointestinal and vascular disorders (hypotension).
Overview of adverse events with onset between the start and end of randomised treatment (safety analysis set; number of events or patients and %).
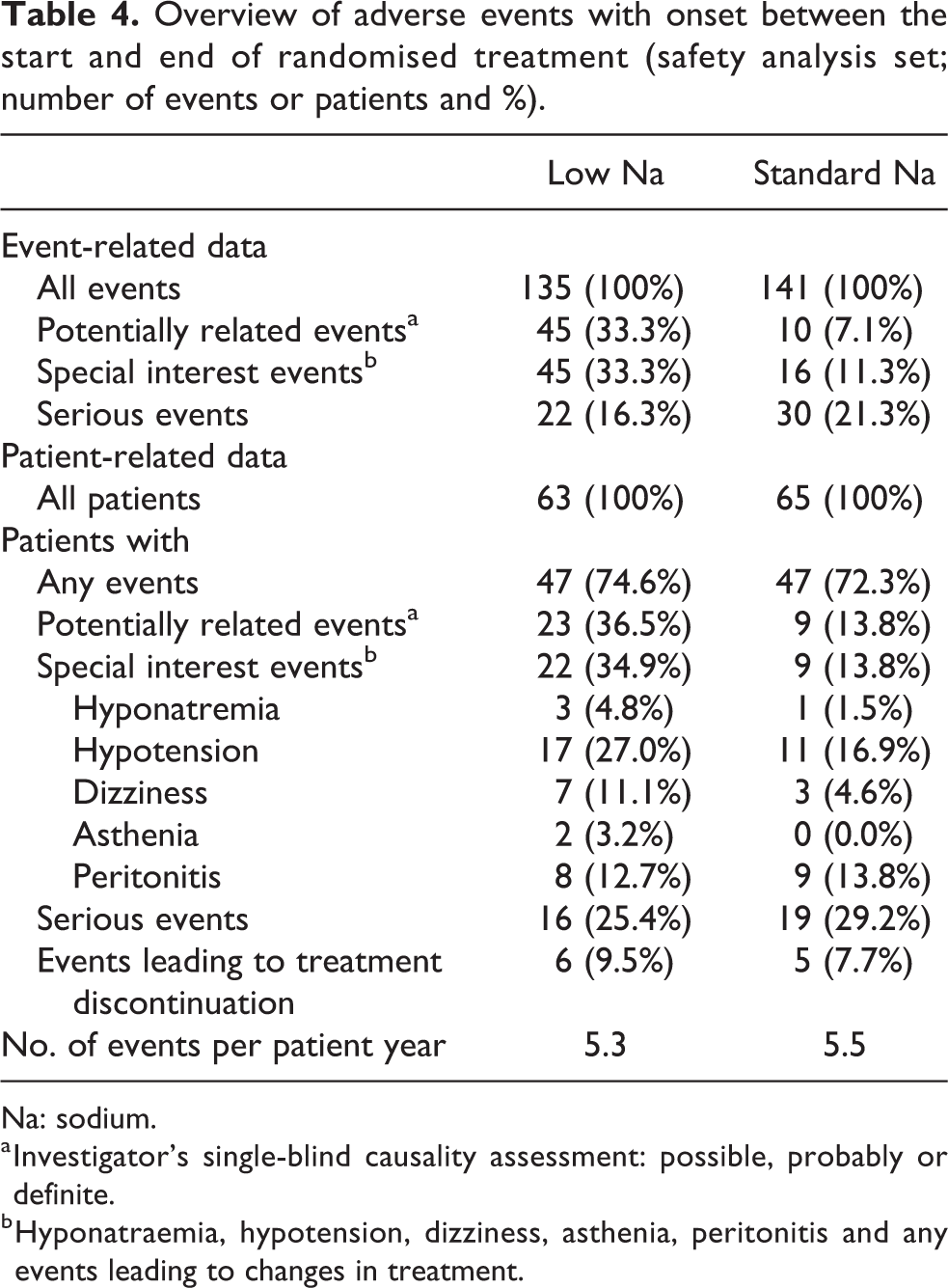
Na: sodium.
a Investigator’s single-blind causality assessment: possible, probably or definite.
b Hyponatraemia, hypotension, dizziness, asthenia, peritonitis and any events leading to changes in treatment.
Among the special interest events, hypotension and dizziness were more common in patients on low-Na treatment than in the standard-Na group, whereas the other event incidences, including hyponatraemia, differed by not more than two affected patients. Three cases of hypotension in the low-Na group and four cases of peritonitis in each group were serious.
With respect to shifts in the mean, there were no noteworthy changes in safety laboratory measures or vital signs, and no important differences between the study groups. Compared to baseline, mean plasma Na in the low-Na group decreased by 1.3 (3.6) and by 1.0 (3.2) mmol/L at 2 and 8 weeks of randomised treatment, respectively, compared to increases by 0.2 (2.7) and 0.2 (2.8) mmol/L in the control group. Between baseline and month 6, decreases by 0.44 (3.73) mmol/L and by 0.50 (2.54) mmol/L were observed for low Na and control, respectively.
There were also no significant differences between both study groups regarding residual renal function, which was measured at baseline, after 2 (p = 0.56) and 6 (p = 0.70) months’ treatment (Table 3).
Discussion
The optimum electrolyte composition of a PD solution is that which best serves the homeostatic needs of the body. 12 Since patients with chronic kidney disease, notably those on dialysis, tend to be fluid and salt overloaded and are thus at an increased risk of cardiovascular morbidity and mortality related to hypertension, 2 –4 the correction of the Na balance must be a primary aim of long-term dialysis treatment. 6 This study investigated whether BP could be lowered by once-daily dwells of a low-Na, glucose-compensated dialysis solution. 11,13,16
For the composite primary end point including a ≥6 mmHg decrease of 24-h SBP or a fall in BP necessitating a medical intervention, superiority of the low-Na solution over standard-Na solution could not be established. With 34.5% and 29.1% for low-Na and standard-Na, respectively, the observed response rates in both groups fell considerably short of the rates of 65% and 40% assumed during sample size planning. The observation that fewer patients in the low-Na group met the criterion of a ≥6 mmHg SBP decrease during ABPM at week 8 is likely explained by the fact that nine patients in this group developed a fall in BP before week 8 that necessitated a clinical decision to reduce antihypertensive medication, which then might have returned the BP to the preintervention level. It is important to note that BP decreases that required a medical intervention were observed only in the low Na group, which might indicate that the single-dwell administration of the 112 mmol/L Na solution led to a sharp BP decrease in some of the subjects, notably in those with an initial 24-h SBP ≤130 mmHg 4 . This observation is consistent with the study’s safety results and one might hypothesise that the investigated PD regimen may lead to undesirable effects, such as hypotension that might result from excessive Na removal, which has been identified as a major mortality risk factor in patients undergoing PD. 17 –20
During the initial 8 weeks of randomised treatment, when antihypertensive medication changes were permitted only in case of hypotension, arterial BP mean values measured with three different methods (24-h ABPM, office measurements, patients’ self-measurements) showed small decreases, little more in the low-Na group as compared to standard-Na. However, the treatment group differences were smaller than anticipated and statistical significance was only found in case of self-measured SBP. BP decreases may have been partly compensated by reductions of antihypertensive medication in hypotensive patients and, after the end of the efficacy period, in other patients as well, when a decrease in the average number of antihypertensive drugs was observed in the low-Na group but not in the standard-Na group.
A potential BP lowering effect of sodium reduced PD solutions might be supported by the observation that patients receiving the low Na solution, unlike those in the control group, showed more pronounced BP decreases during nocturnal readings, that is, following the low-Na exchange, than during the day, an effect that was also reported by Davies et al. using a similar treatment regimen. 13 It is also worth mentioning in this context that significant advantages for the low Na solution were observed in self-measured BP, the only BP assessment performed under blinded conditions. There is evidence that patients’ BP self-measurements may offer advantages over routine dialysis unit measurements for determining cardiovascular risk. 21,22
An analysis of the effect of a low-Na solution specially in APD patients would have been desirable since the Na sieving phenomenon 23,24 would favour the use of Na-reduced solutions. However, since, overall, only 24% of our patients were on APD and were not well balanced between the two groups, an analysis on this subgroup was not feasible.
The solution concept in our study follows that applied in one arm of the study by Davies et al., 13 using once daily a PD fluid with the Na concentration reduced to 115 mmol/L, and an increased glucose concentration to compensate for loss of osmolality. The major finding regarding effect on BP was the significant reduction of night-time mean arterial pressure after 2 months of exposure, which was similarly observed in our study. In contrast to this, the treatment regimen in the study by Rutkowski et al. contained a PD solution, which was only moderately reduced in Na concentration (125 mmol/L), and not glucose compensated. 25 Here, a trend of different decrease of BP in favour of the low Na group was observed in the entire study population, whereas a post hoc subgroup analysis revealed a significant difference in favour of the low-Na solution for SBP and DBP for patients with a GFR <6 mL/min/1.73 m2. 26 It is of note that a higher frequency of hypotension has been observed with the once per day application of a very low-sodium PD solution both in our study and in that by Davies et al. 13 than with the continuous application of a PD solution with moderately reduced sodium concentration. 25 Thus, a more gradual introduction of the low-Na solution could improve tolerance to achieve BP decreases without an appreciable risk of hypotensive episodes. 25,26
The results raise some questions regarding the appropriateness of the composite primary end point, one of whose efficacy criteria was actually an important adverse reaction in PD (hypotension). The fact that the two efficacy criteria included in the primary end point were competitive (patients who required a reduction of antihypertensive medication were counted for this criterion even if they also showed a ≥6 mmHg SBP decrease) complicated the interpretation of the results. Given that clinicians were not blinded to the intervention, it is possible that the need to reduce antihypertensive medication represents performance bias. Moreover, the reduction of antihypertensive medication likely counteracted the BP decrease induced by Na removal and thus should probably be included into future investigations as an efficacy criterion. Another limiting factor is that neither Na sensitivity and other predispositions nor nutritional factors (e.g. salt intake and thirst) were assessed.
Conclusions
In conclusion, superiority of the low-Na solution over standard-Na solution in BP control could not be confirmed. Some results of this study indicate to a potential of low-Na PD solutions for improving BP while reducing the antihypertensive treatment burden. Although the 112 mmol/L low-Na solution was generally well tolerated and showed a safety profile similar to that of the standard Na solution, the single-dwell administration was associated with an increased risk of hypotension. Therefore, clinical development should aim at the solution composition, optimal sodium concentration, prescription and target patients likely to benefit from a Na-reduced PD solution.
Supplemental material
Supplemental Material, 924136_supp_mat - Single-dwell treatment with a low-sodium solution in hypertensive peritoneal dialysis patients
Supplemental Material, 924136_supp_mat for Single-dwell treatment with a low-sodium solution in hypertensive peritoneal dialysis patients by Simon Davies, Börje Haraldsson, François Vrtovsnik, Vedat Schwenger, Stanley Fan, Alexandre Klein, Saynab Atiye and Adelheid Gauly in Peritoneal Dialysis International
Footnotes
Acknowledgements
We acknowledge the commitment and dedication of the investigators and their clinical teams at the participating centers during the trial: BH, Sahlgrenska University Hospital Gothenburg, Sweden; OS, N Clyne, University Hospital Lund, Sweden; A-C Johansson, University Hospital Malmö, Sweden; H Hadimeri, Skövde Hospital, Sweden; F-D Nielsen, Södra Älvsborgs Sjukhus Borås, Sweden; P Dahlberg, Nord Älvsborg County Hospital, Trollhättan, Sweden; O Heimburger, Karolinska University Hospital Stockholm, Sweden; H Furuland, Uppsala University Hospital, Sweden; T Elung-Jensen, Rigshospitalet, Copenhagen, Denmark; P Rössel, Aalborg Hospital, Denmark; E Randers, Viborg Hospital, Denmark; FV, Bichat—Claude Bernard Hospital Paris, France; B Morel, Chambéry Hospital, France; B Faller, AK, Hospital Pasteur Colmar, France; F Heibel, University Hospital, Strasbourg, France; J-P Ryckelynck, University Hospital Center, Caen, France; C Courivaud, C Bresson-Vautrin, CHRU—Hospital Saint-Jacques, Besançon, France; S Lavaud, E Canivet, ARPDD Reims, France; A Caillette-Baudoin, CALYDIAL Vienne, Irigny, France; VS, University Hospital Heidelberg, Germany; M Haag-Weber, St Elisabeth Hospital, Straubing, Germany; R Brunkhorst, Hospital Oststadt-Heidehaus, Hannover, Germany; M Nebel, B Kitsche, KFH-Nierenzentrum Köln-Merheim, Germany; I Quack, University Hospital Düsseldorf, Germany; S Nunnenkamp, KFH-Nierenzentrum Passau, Germany; S Ludewig, KFH-Nierenzentrum Eberswalde, Germany; S Weiner, N Wirtz, Krankenhaus der Barmherzigen Brüder Trier, Germany; C Hugo, S Parmentier, University Hospital Dresden, Germany; T Schneider, Internistische Gemeinschaftspraxis—Nephrologisches Zentrum Stuttgart, Germany; SD, University Hospital of North Staffordshire, UK; SF, London Royal Barts Hospital, UK; S Ramakrishna, Royal Shrewsbury Hospital, UK; KS Sandhu, New Cross Hospital Wolverhampton, UK; I Dasgupta, Heartlands Hospital Birmingham, UK; U Udayaraj, Southmead Hospital, Bristol; R Asghar, Royal Preston Hospital, UK; A Davenport, London Royal Free Hospital, UK. The authors would like to thank B Rippe, Department of Nephrology, University Hospital Lund, Sweden, recently deceased, for his scientific contribution and advice to the design and performance of the study as part of the Steering Committee. Furthermore, the support from the following persons being part of the Data Safety Monitoring Board is gratefully acknowledged: A Slingeneyer, Montpellier, France; M Lichodziejewska-Niemierko, Department of Nephrology Medical University Gdańsk, Poland; S Konings, Catharina Ziekenhuis Eindhoven, the Netherlands; G Bobrie, Hypertension Unit of Hôpital Européen Georges Pompidou, Paris, France.
Author contributions
SD, BH, FV and VD were members of the Steering Committee of the study and contributed to the development of the project, the design and scientific conduct of the study. SD, BH, FV, VS, SF, AK, SA and AG supported the interpretation of the data, the preparation, review and approval of the manuscript. Special thanks to Andreas Völp, Psy Consult Scientific Services, Frankfurt, Germany, for his contribution to the final analysis and the compilation of the manuscript.
Declaration of conflicting interests
The author(s) declared the following conflicts of interest with regards to the research, authorship, and/or publication of this article: FV has received lecture fees and travel funding to research meetings from Alexion, Amgen, Baxter and Fresenius Medical Care. SF received consulting, lecture fees, and travel funding by Baxter and Vifor. VS received lecture fees from Fresenius Medical Care. SD has received research funding and advisory committee fees from Baxter HealthCare. AG and SA are full-time employees at Fresenius Medical Care. The other authors declare that they have no other relevant financial interests.
Ethical approval
The trial was conducted in 30 centres in Sweden, France, Germany, Denmark, and the United Kingdom, enrolling patients between October 2008 and February 2014. The study protocol and its attachments and amendments were reviewed and approved by the competent independent ethics committees in the participating countries.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The clinical trial was initially funded by Gambro AB and later by Fresenius Medical Care.
Informed consent to participate
All patients provided written informed consent. The principles of Good Clinical Practice and the Declaration of Helsinki were adhered to.
Informed consent to publish
Written informed consent was obtained from the patient(s) for their anonymised and aggregated information to be published in this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.