
Abstract
The effect of a polymeric plasticizer on crosslinking behavior and performance of nitrile butadiene rubber (NBR, SKN-40) was evaluated under thermal, radiation, and thermoradiation vulcanization. Under thermoradiation curing (250 kGy +150°C) at low sulfur content (0.2 phr), the polymeric plasticizer produces a markedly higher effective crosslink density than non-plasticized systems, demonstrating a synergistic interaction between sulfur-mediated and radiation-induced crosslinking. FTIR analysis shows enhanced consumption of butadiene unsaturation (∼965–970 cm−1) without alteration of nitrile groups, indicating selective network formation. EPR spectroscopy reveals rapid recombination of radiation-induced macroradicals, with stabilization of weak carbon-centered radicals (g = 2.0031, ΔH = 0.44 mT) within 48 h, confirming suppression of post-irradiation chain scission. As a result, polymeric plasticizer-assisted thermoradiation vulcanizates exhibit the highest intrinsic viscosity (∼3.0 dL g−1), tensile stress at 300% elongation (M300) (∼10 MPa), and tear resistance (∼27 MPa). Thermogravimetric analysis demonstrates delayed thermal degradation (T5% ≈ 390°C, T10% ≈ 420°C) and increased char residue (∼48%), consistent with the formation of a compact hybrid network combining thermally stable carbon–carbon crosslinks with short sulfur bridges.
Keywords
Introduction
Nitrile butadiene rubber (NBR) is one of the most widely used polar elastomers for applications that require resistance to oils, fuels, and aggressive chemical environments, such as seals, hoses, and flexible technical components. In recent years, research has shown that the performance and durability of NBR-based materials are determined not only by crosslink density, but also by the chemical nature, spatial distribution, and stability of crosslinks within the polymer network.1–3 As a result, network topology has become an important design parameter for improving long-term mechanical performance and thermal resistance.
Conventional sulfur vulcanization mainly produces polysulfidic and disulfidic crosslinks (–C–Sx–C–). These bonds provide good elasticity, but their thermal stability is limited due to bond rearrangement and scission at elevated temperatures. 4 Several studies have reported that sulfur-cured NBR undergoes gradual degradation during prolonged thermal or oxidative exposure, leading to a reduction in mechanical strength and dimensional stability. 5 In contrast, radiation-induced vulcanization primarily forms carbon–carbon (–C–C–) crosslinks through radical recombination along the polymer backbone, resulting in improved thermal and chemical resistance.6,7 However, radiation curing is a competitive process in which crosslinking and chain scission occur simultaneously, often restricting the achievable mechanical properties unless radical reactions are effectively controlled. 8
To overcome the limitations of single curing methods, hybrid approaches combining thermal and radiation curing have attracted increasing attention. Recent studies indicate that the efficiency of such hybrid systems strongly depends on formulation components that influence radical mobility and recombination during irradiation.9–11 Despite this potential, thermoradiation curing of NBR has not yet been sufficiently studied from a mechanistic point of view. One factor that has received limited attention is the role of plasticizers under radiation conditions. Plasticizers are commonly used to improve flexibility and processability and are often considered inert additives. However, recent studies in polymer physics and radiation chemistry suggest that plasticizers—especially polymeric ones—can significantly affect segmental mobility, free volume, and radical diffusion within polymer networks.12–16 Under irradiation, increased chain mobility may reduce irreversible chain scission and promote radical recombination, thereby influencing crosslink structure and post-curing stabilization. 17 Nevertheless, systematic studies on the effects of polymeric plasticizers in thermoradiation-cured NBR systems remain scarce.
Most studies on NBR curing rely mainly on mechanical testing and swelling measurements to evaluate network structure. While useful, these methods provide limited information about radical processes and post-curing reactions. Electron paramagnetic resonance (EPR) spectroscopy allows direct observation of radical formation, stabilization, and recombination in irradiated polymers and has proven to be a powerful tool for studying radiation-induced mechanisms.18–20 However, EPR has rarely been applied to hybrid curing systems involving sulfur, radiation sensitizers, and plasticizers, leaving important mechanistic aspects unresolved.
In this study, we investigate the role of polymeric plasticizers in network formation of NBR cured by thermal, radiation, and combined thermoradiation methods. Sulfur vulcanization, γ-irradiation, and their combination were carried out in the presence of dimethylphenylmaleimide (DMPM) and ZnO to clarify how polymeric plasticizers influence crosslink topology, radical stabilization, and thermal behavior. Sol–gel analysis, FTIR spectroscopy, EPR, differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and mechanical testing were used to establish structure–property relationships in hybrid NBR networks. The results show that polymeric plasticizer-assisted thermoradiation curing leads to compact networks that combine thermally stable carbon–carbon crosslinks with flexible sulfur bridges, offering an effective approach for developing high-performance NBR materials for demanding thermal and chemical applications.
Experimental part
Materials
Nitrile butadiene rubber (NBR, grade SKN-40) was used as the elastomer matrix. The material was supplied by Guclu Polymer LLC (Istanbul, Türkiye) and selected due to its medium acrylonitrile content, which provides an optimal balance between oil resistance and processability, making it representative of typical industrial NBR formulations.
Rubber compounds were formulated using a conventional sulfur-based curing system combined with radiation-active additives. Sulfur (2.0 phr) was employed as the primary vulcanizing agent, while 2-mercaptobenzothiazole (MBT, Captax, 1.5 phr) served as the accelerator. Zinc oxide (ZnO, 5.0 phr) was used as an activator to enhance curing efficiency. Dimethylphenylmaleimide (DMPM, 1.5 phr) was incorporated as a radiation-chemical sensitizer to promote radical-mediated crosslinking under γ-irradiation. Carbon black P324 (40 phr) was used as a reinforcing filler to provide mechanical strength and structural integrity.
A polymeric plasticizer was introduced at a concentration of 2.5 phr. The plasticizer consisted of industrial oil U-61 and polyvinyl alcohol (PVA) as the polymeric component. PVA was selected due to its polarity and compatibility with NBR. 21 The polymeric plasticizer was prepared under laboratory conditions by blending U-61 oil with 2.5 wt.% PVA, yielding a homogeneous polymer–oil mixture.
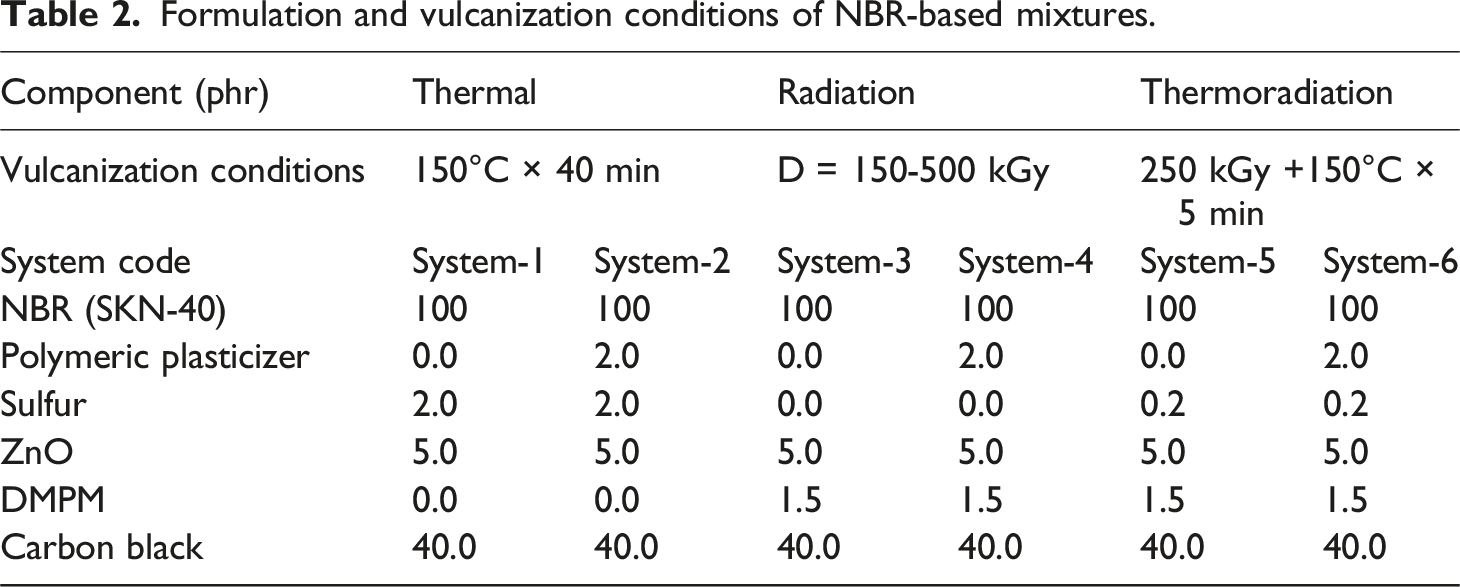
Summarizes the formulation of the NBR-based elastomer compounds.

Preparation and characterisation of rubber compounds
Mechanical plasticization and compounding of NBR were performed using a laboratory two-roll mill (roll dimensions: 160 × 320 mm; model LB-160, Shanghai Rubber Machinery Co., Ltd, China). Mixing was carried out at temperatures between 30 and 70°C with a friction ratio of 1:2 and a batch mass of approximately 100 g. Components were added sequentially according to Table 1, and each formulation was mixed for 15 min to ensure homogeneous dispersion.
Thermal vulcanization was conducted using an electrically heated hydraulic press (QLB-D400 × 400, Qingdao Xincheng Yiming Rubber Machinery Co., Ltd, China) at 150°C for 40 min.
Radiation vulcanization was performed by exposing the samples to γ-irradiation from a 60Co source at a dose rate of 6.4 Gy s−1, with absorbed doses ranging from 150 to 500 kGy. For radiation-cured systems, DMPM was used to enhance radiation-induced crosslinking and suppress excessive chain scission.
Thermoradiation vulcanization combined moderate thermal treatment (150°C for 5 min) with γ-irradiation at an absorbed dose of 250 kGy, enabling the simultaneous formation of sulfur-based and carbon–carbon crosslinks.
Formulation and vulcanization conditions of NBR-based mixtures.
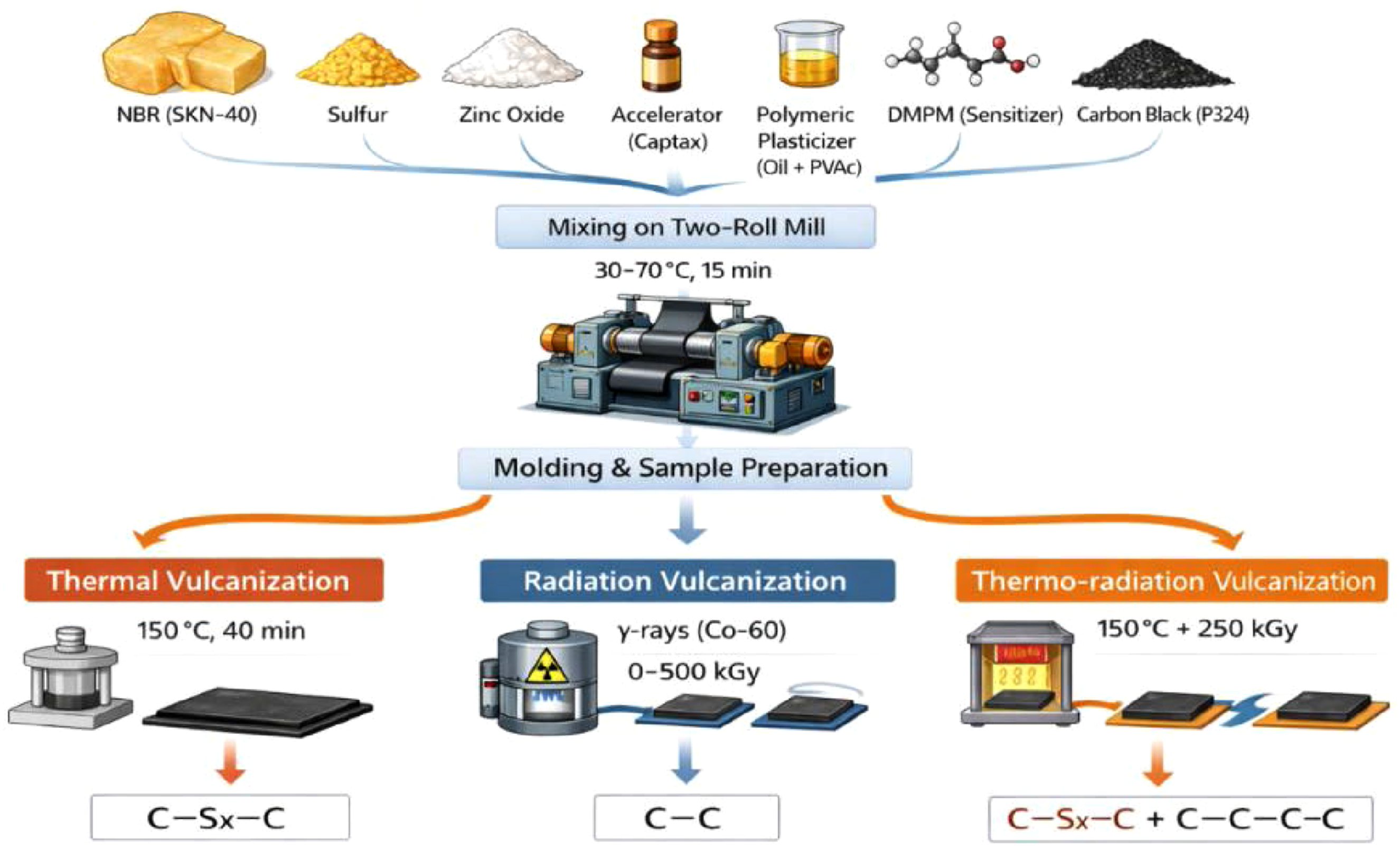
Preparation and vulcanization routes of NBR-based elastomer compounds.
Gel content and cross-linking density by the sol–gel analysis
The degree of crosslinking and equilibrium swelling behavior were determined by sol–gel analysis using analytical-grade toluene (Sigma-Aldrich, Germany). Crosslink density was calculated from equilibrium swelling data using the Flory–Rehner equation.22,23
FTIR analysis
FTIR spectra were recorded in the range of 4000–500 cm−1 using a Varian 640 FTIR spectrometer (Agilent Technologies, USA) to monitor chemical changes associated with network formation. Radical formation and stabilization during radiation and thermoradiation vulcanization were investigated by electron paramagnetic resonance (EPR) spectroscopy using a Bruker EMXmicro X-band spectrometer (Bruker BioSpin GmbH, Germany) at room temperature.24,25
Intrinsic viscosity measurements
Intrinsic viscosity measurements were performed in toluene at 20°C using an Ubbelohde capillary viscometer (Schott Instruments, Germany), and molecular weight changes were estimated via the Mark–Houwink relationship for the NBR–toluene system.4,5
Mechanical testing
Mechanical properties were evaluated by uniaxial tensile testing using a CMT-20 universal testing machine (Liangong Testing Technology Co., Ltd, Jinan, China) equipped with a clip-on extensometer. Rectangular specimens with dimensions of 70 mm × 12.5 mm × 3.5 mm were tested. Mooney viscosity of uncured compounds was measured at 100°C using a Mooney viscometer (MV 2000, Alpha Technologies, USA).
Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) analyses
Thermal properties were assessed by differential scanning calorimetry (DSC) using a NETZSCH DSC 214 Polyma under nitrogen atmosphere to determine glass transition temperatures. Thermogravimetric analysis (TGA) was performed using a NETZSCH TG 209 F3 Tarsus to evaluate thermal stability and degradation behavior.
EPR spectroscopy analysis
EPR measurements were carried out on a Bruker EMX microX spectrometer operating in the X-frequency range, with a modulation frequency of 9.8 · 109 Hz, (λ = 3 cm). Samples were placed in sealed quartz ampoules with a diameter of 3 mm and a length of 15–20 cm, and then the ampoules were placed in the equipment for research. The concentration of paramagnetic centers and g factors were determined and compared with the reference data by the method described in literature.26,27
Results and discussion
Influence of vulcanization pathway and polymeric plasticizer on crosslinking behavior of NBR
The formation of a three-dimensional network in nitrile butadiene rubber (NBR) depends on the curing mechanism and on factors that influence molecular mobility during crosslinking. In this study, crosslink formation was assessed indirectly through equilibrium swelling measurements and sol–gel analysis, which provide information on the effective crosslink density of the resulting vulcanizates.
During conventional sulfur vulcanization, crosslinks are formed through sulfur bridges connecting unsaturated segments of the NBR chains. In the system without polymeric plasticizer (System-1), the increase in effective crosslink density with curing time corresponds to the progressive formation of polysulfidic and disulfidic linkages typical of sulfur-cured NBR (Figure 2.). When the polymeric plasticizer is added (System-2), higher effective crosslink densities are obtained at comparable curing times. This behavior indicates that the presence of the plasticizer improves the accessibility of reactive sites during vulcanization, leading to a more complete incorporation of sulfur into the network, rather than changing the chemical nature of sulfur crosslinks. Effect of the polymeric plasticizer on the degree of crosslinking of NBR during thermal sulfur vulcanization as a function of curing time (System-1; System-2).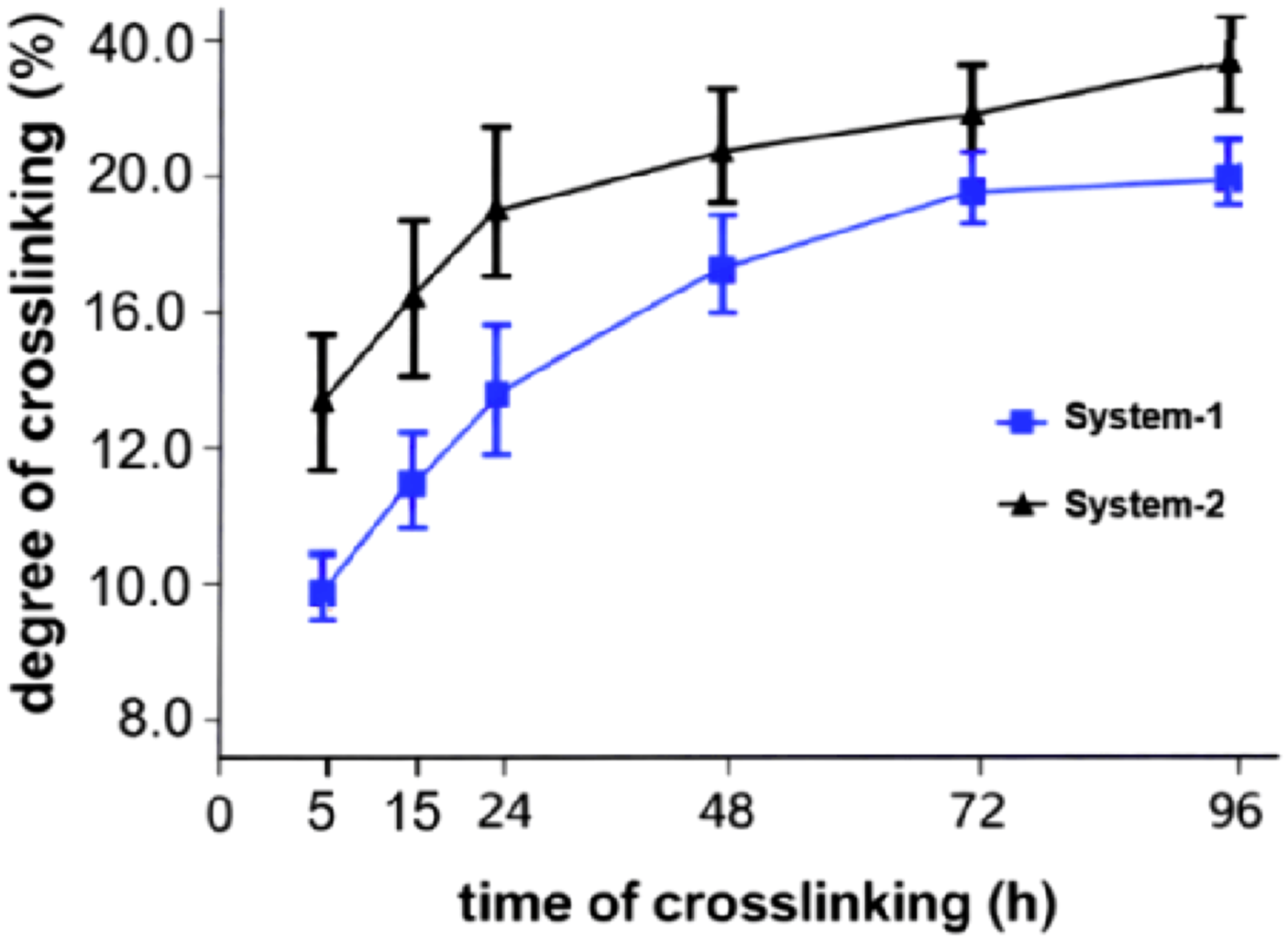
Radiation vulcanization leads to a fundamentally different type of network. In sulfur-free systems exposed to γ-irradiation (System-3), crosslink formation occurs predominantly via radical recombination along the polymer backbone, resulting in carbon–carbon crosslinks. However, this process competes with radiation-induced chain scission, which limits the net increase in effective crosslink density. The introduction of the polymeric plasticizer (System-4) results in higher crosslink densities at the same absorbed doses, suggesting that radical recombination is favored relative to chain scission. In this case, the plasticizer does not act as a crosslinking agent but modifies the physical environment of the polymer chains, facilitating encounters between macroradicals.
The effect of the polymeric plasticizer is most pronounced under thermoradiation vulcanization, where sulfur curing and γ-irradiation are combined. At low sulfur content (0.2 phr), the PP-free system (System-5) shows limited crosslink formation, indicating inefficient utilization of both sulfur- and radiation-induced reactions. In contrast, the polymeric plasticizer containing system (System-6) exhibits a substantially higher effective crosslink density under identical conditions. This result suggests that the polymeric plasticizer promotes the interaction between radiation-generated macroradicals and sulfur species during the subsequent thermal step, leading to more efficient incorporation of both carbon–carbon and sulfur crosslinks into the network.
These observations indicate that differences between the curing routes are not only reflected in the amount of crosslinking but also in the nature of the resulting network. Sulfur vulcanization produces networks dominated by sulfur bridges of varying length, while radiation vulcanization yields networks primarily stabilized by carbon–carbon crosslinks. Thermoradiation vulcanization in the presence of the polymeric plasticizer leads to a mixed crosslink structure, in which a carbon–carbon crosslinked backbone is supplemented by short sulfur bridges. This hybrid network structure provides a rational explanation for the enhanced mechanical strength and thermal stability discussed in subsequent sections.
Overall, the polymeric plasticizer influences crosslink formation by modifying the physical conditions under which curing reactions occur. Its effect is limited in single-route curing but becomes critical when sulfur vulcanization and radiation crosslinking are combined, enabling the formation of a more efficiently crosslinked and structurally integrated NBR network.
This enhancement indicates that increased segmental mobility facilitates more effective participation of sulfur in network formation, leading to a denser and more homogeneous spatial network.28–32 Radiation-cured systems exhibit a markedly different trend (Figure 3). In samples cured by γ-irradiation without polymeric plasticizer (System-3), the degree of crosslinking increases only moderately with absorbed dose, reflecting the competitive balance between radical-induced crosslinking and chain scission processes typical for irradiated elastomers.8,33 In contrast, the presence of polymeric plasticizer (System-4) leads to a pronounced increase in crosslinking efficiency across the entire dose range. This behavior suggests that the polymeric plasticizer promotes radical diffusion and recombination, thereby shifting the radiation-chemical balance toward network formation rather than molecular degradation.
34
Effect of the polymeric plasticizer (PP) on the degree of crosslinking of NBR under γ-radiation vulcanization as a function of absorbed dose (System-3: System-4: NBR + PP + DMPM).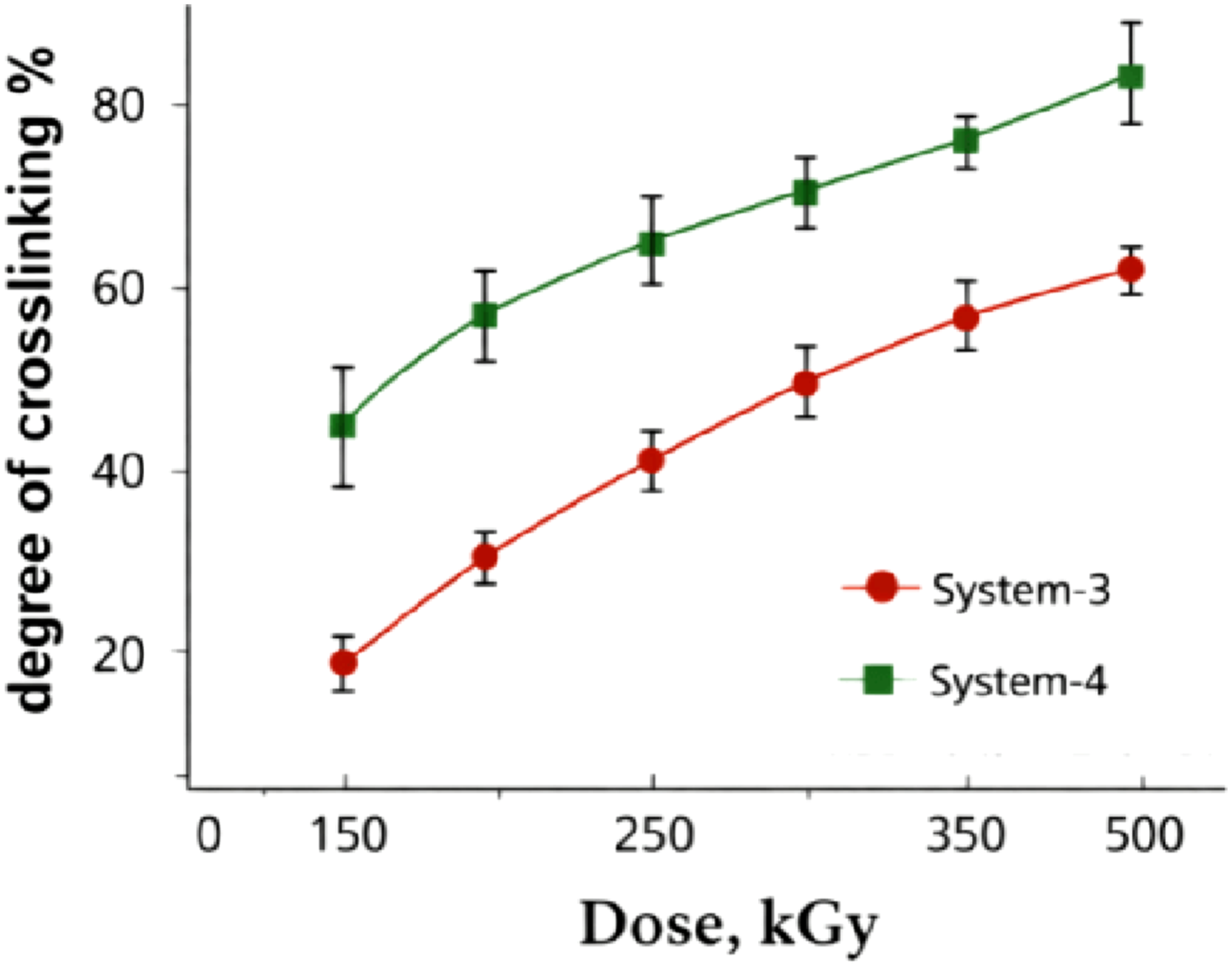
The most significant effect of the polymeric plasticizer is observed under thermoradiation curing conditions (Figure 4), where sulfur vulcanization and γ-irradiation act simultaneously. At low sulfur content (0.2 phr), systems without polymeric plasticizer (System-5) show only a limited increase in crosslinking degree with absorbed dose. In contrast, the polymeric plasticizer containing system (System-6) exhibits a substantial enhancement of crosslinking. This result demonstrates a clear synergistic interaction between sulfur-mediated crosslink formation and radiation-induced carbon–carbon bond generation, enabled by improved chain mobility in the presence of the polymeric plasticizer. Influence of the polymeric plasticizer on the degree of crosslinking of NBR (SKN-40) under thermoradiation vulcanization at low sulfur content (0.2 phr), comparing systems without polymeric plasticizer (System-5) and with polymeric plasticizer (System-6).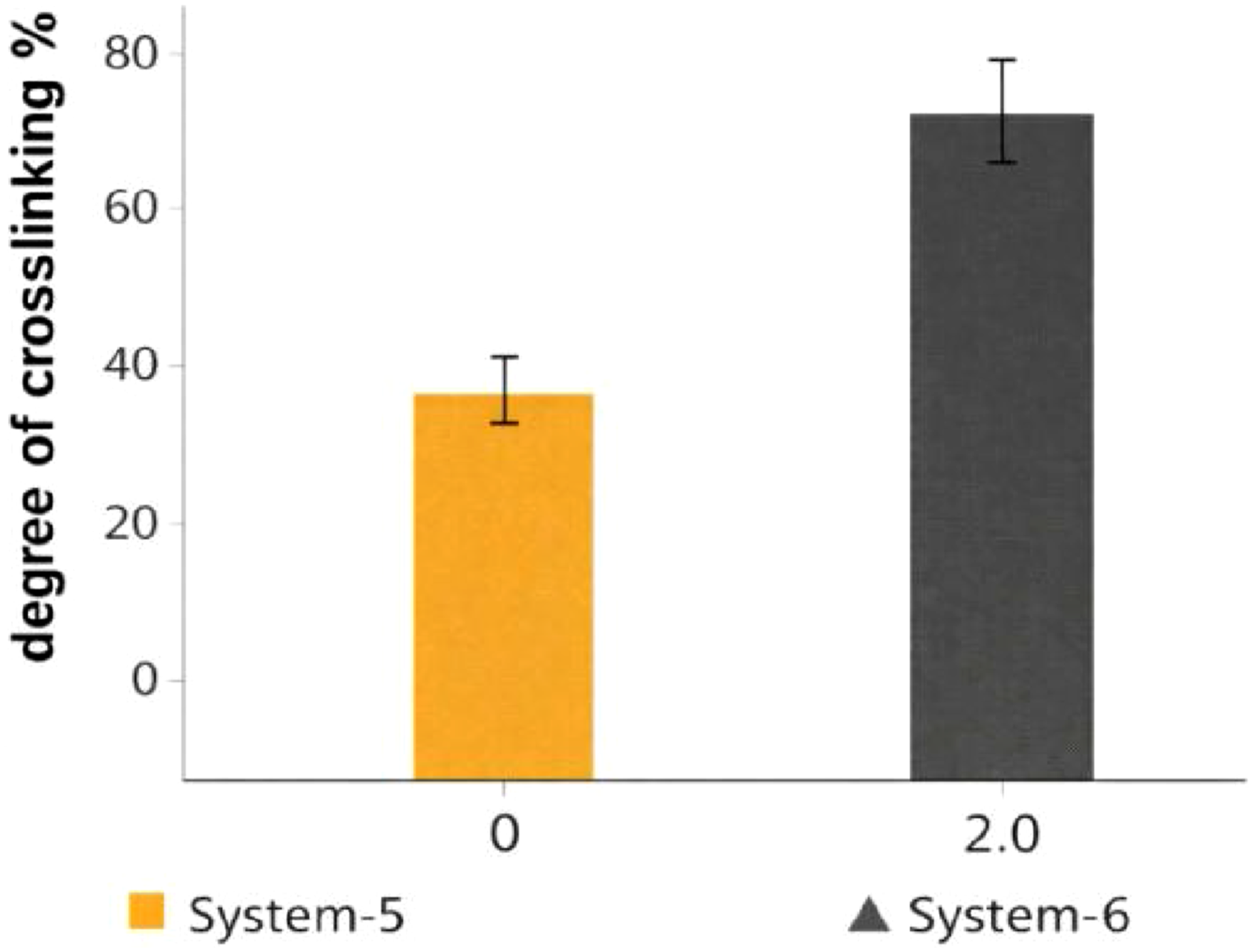
It is important to note that the observed differences are not solely quantitative but also reflect qualitative changes in network architecture. While sulfur-cured systems are dominated by polysulfidic crosslinks, radiation curing favors thermally stable carbon–carbon bonds. Thermoradiation curing in the presence of polymeric plasticizer results in a hybrid network combining strong C–C linkages with flexible sulfur bridges. Such a network architecture provides an optimal balance between rigidity and elasticity and forms the structural basis for the enhanced mechanical and thermal performance discussed in subsequent sections.35–37
Overall, these results demonstrate that the polymeric plasticizer plays an active and decisive role in regulating crosslinking efficiency and network homogeneity. Its influence is most pronounced under thermoradiation conditions, where it enables effective coupling of thermal and radiation-induced crosslinking mechanisms, leading to the formation of compact and well-developed NBR networks.
FTIR analysis of network formation in NBR systems (Systems 1–6)
Figure 5 shows the FTIR spectrum of the polymeric plasticizer. A broad absorption band at 3200–3400 cm−1 corresponds to –OH stretching vibrations of PVA, while strong aliphatic C–H stretching bands at approximately 2920 and 2850 cm−1 originate from the oil phase. Due to the low PVA content, characteristic PVA bands in the 1200–900 cm−1 region partially overlap with oil-related vibrations, resulting in broadened composite features. This formulation combines the softening effect of the oil with the macromolecular character of the polymeric component, enabling controlled modification of chain mobility during vulcanization. FTIR spectrum of the polymeric plasticizer.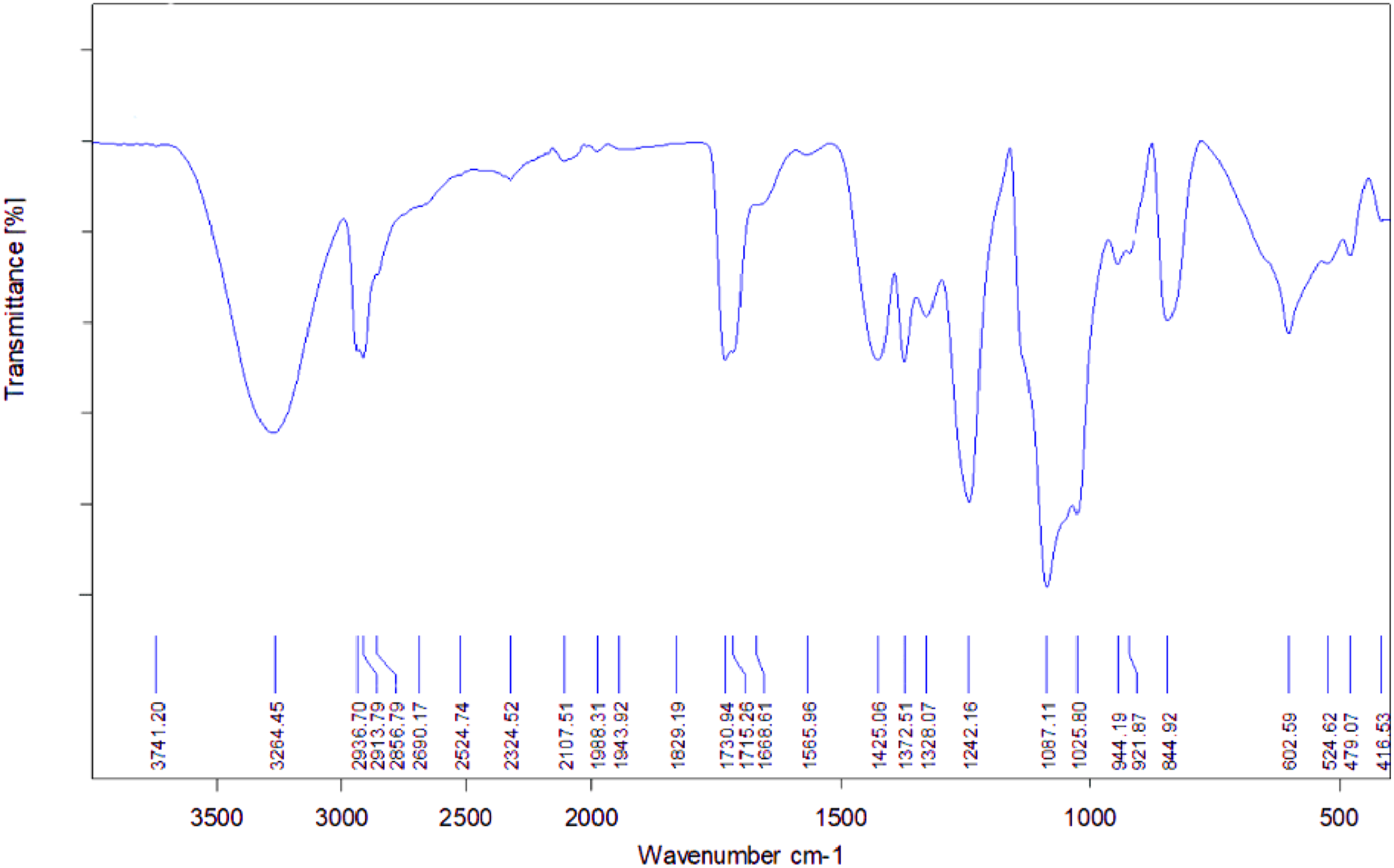
Figure 6. Presents the FTIR spectra of representative NBR compounds cured via three different vulcanization routes: conventional thermal curing (System-1), radiation-induced curing (System-3), and combined thermoradiation curing (Systems-5 and -6). Although these systems differ in curing chemistry, sulfur content, and the presence of a polymeric plasticizer (PP), all spectra retain the characteristic infrared fingerprint of nitrile butadiene rubber. This observation confirms that the applied curing strategies restructure the elastomeric network while preserving the fundamental chemical identity of the NBR matrix within the detection limits of FTIR spectroscopy. FTIR spectra of thermoradiation vulcanizate NBR + DMPM + PP + Sulfur + ZnO.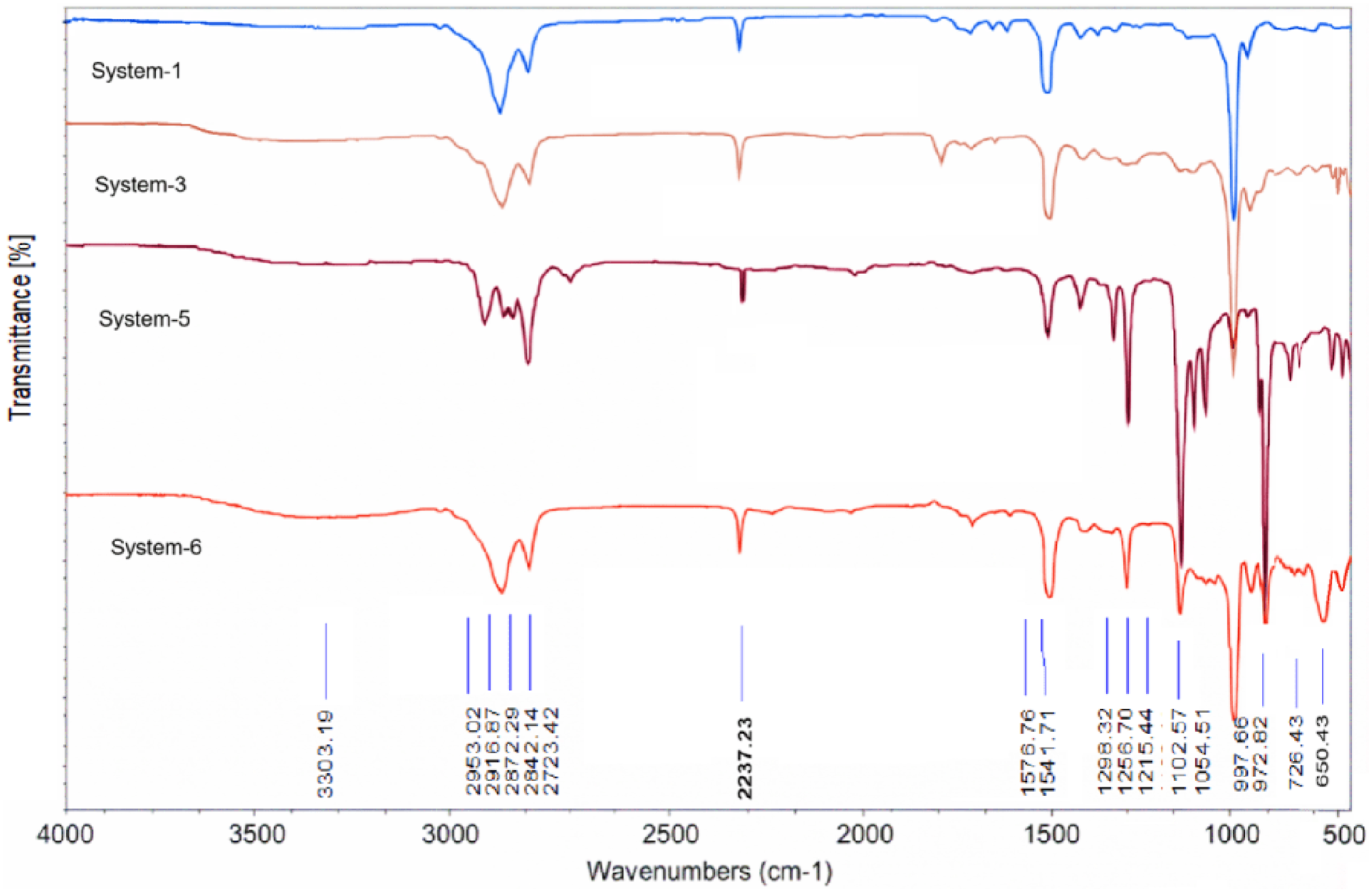
All investigated systems exhibit a strong absorption band at ∼2235–2240 cm−1, which is attributed to the stretching vibration of the nitrile group (–C≡N), a defining structural feature of NBR. The position and relative intensity of this band remain essentially unchanged across thermal, radiation, and thermoradiation curing regimes. This invariance indicates that the polar nitrile functionality does not participate directly in the crosslinking reactions and remains chemically stable under the applied processing conditions. In addition, the aliphatic –CH stretching bands in the 2920–2850 cm−1 region are preserved in all samples, reflecting the retention of the hydrocarbon backbone. Together, these features demonstrate that network formation proceeds selectively through reactions involving unsaturated segments rather than through degradation or chemical transformation of the main polymer chain.
The most sensitive FTIR indicator of crosslink development in NBR appears in the ∼965–970 cm−1 region, which is associated with vibrations of butadiene-derived unsaturated units. A systematic decrease in the intensity of this band is observed when moving from the thermally cured system to the radiation- and thermoradiation-treated samples. This progressive attenuation reflects the consumption of C = C sites during network formation and provides direct spectroscopic evidence of increasing structural conversion. Importantly, this trend is fully consistent with the independently determined sol–gel fractions and crosslink density values, supporting the interpretation that higher curing efficiency corresponds to a greater depletion of unsaturated units. 26
Thermal vulcanization (System-1): Sulfur-controlled network formation
System-1 represents a conventional sulfur-cured NBR formulation containing 2.0 phr sulfur and no radiation sensitizer. Under thermal vulcanization, crosslinking proceeds predominantly via the formation of sulfur bridges (–C–Sx–C–). Because sulfur-containing bonds exhibit weak infrared activity, their formation is not manifested as sharp diagnostic peaks but rather as subtle changes within the fingerprint region (1200–800 cm−1), with possible weak contributions extending into the 650–550 cm−1 range. Consequently, the FTIR signature of System-1 is characterized mainly by partial suppression of the ∼965–970 cm−1 band, while the overall spectral profile remains close to that of uncured NBR. This behavior is typical of elastomer networks dominated by sulfur crosslinks.
Radiation vulcanization (System-3): Radical-driven C–C crosslinking
In contrast, System-3 contains no sulfur and is cured exclusively by γ-irradiation in the presence of dimethylphenylmaleimide (DMPM) and ZnO. Under these conditions, network formation is governed by radiation-induced radical generation along the polymer chains followed by recombination reactions, leading predominantly to carbon–carbon crosslinks. The FTIR spectrum of System-3 shows a more pronounced reduction of the unsaturation-related band near ∼965–970 cm−1 compared with the thermally cured system, indicating a higher extent of double-bond conversion. The absence of sulfur eliminates contributions in sulfur-sensitive low-wavenumber regions, further supporting the formation of a C–C-dominated network structure consistent with a radiation-driven crosslinking mechanism enhanced by the maleimide co-agent.
Thermoradiation vulcanization (Systems-5 and -6): Formation of mixed networks
Systems-5 and -6 are cured under combined thermoradiation conditions (250 kGy followed by a short thermal treatment at 150°C) and contain both DMPM and a low sulfur concentration (0.2 phr). This formulation strategy is designed to generate a hybrid network in which radiation-induced C–C crosslinks form the primary structural framework, while the limited sulfur content contributes a secondary population of short sulfur bridges during the thermal step. In Figure 7, both thermoradiation-cured samples exhibit a stronger suppression of the ∼965–970 cm−1 band than System-1, indicating a higher extent of unsaturation consumption. At the same time, weak spectral features in the 650–550 cm−1 region are consistent with the presence of sulfur-derived linkages, even at low sulfur loading. These observations confirm the formation of a mixed crosslinking architecture rather than a purely sulfur- or radiation-dominated network. EPR spectrum of a thermoradiation sample NBR + DMPM + PP + Sulfur + ZnO; (a) after vulcanization; (b) after 48 h.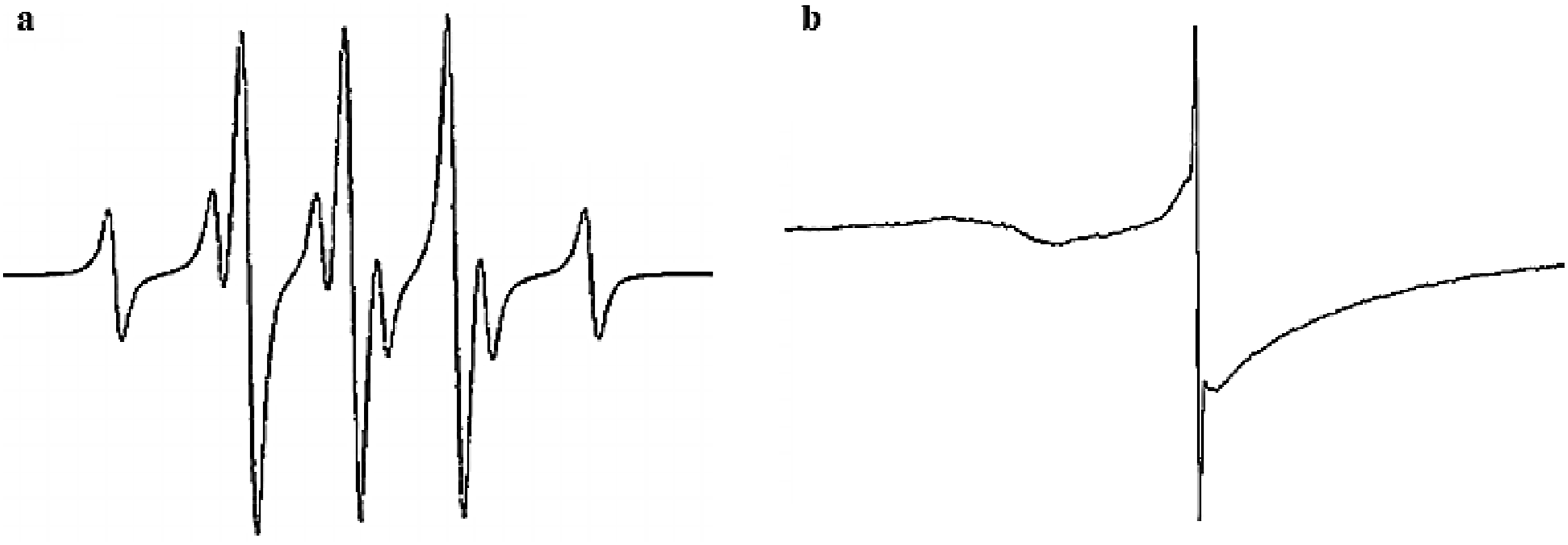
Effect of the polymeric plasticizer under thermoradiation conditions
A direct comparison between Systems-5 (PP = 0) and −6 (PP = (2) isolates the effect of the polymeric plasticizer, since all other formulation parameters and curing conditions are identical. The FTIR spectrum of System-6 shows a more pronounced reduction in the unsaturation-related band compared with System-5, indicating a higher effective extent of unsaturation consumption and network development. This effect can be attributed to enhanced chain mobility induced by the polymeric plasticizer, which facilitates radical diffusion and recombination during irradiation. Moreover, increased segmental mobility improves the efficiency of the brief thermal step, allowing the low sulfur content to participate more uniformly in network formation. 27 As a result, the plasticized system develops a more homogeneous and effectively crosslinked hybrid network within the same curing window.
Collectively, the FTIR results demonstrate that all curing strategies preserve the chemical integrity of the NBR backbone, as evidenced by the stable nitrile absorption. Network formation is primarily reflected by the progressive consumption of unsaturated units, with both the extent and dominant mechanism strongly dependent on the curing route. Thermal curing yields a sulfur-bridged network, radiation curing promotes carbon–carbon crosslinking, and thermoradiation curing produces a hybrid structure combining both mechanisms. Within the thermoradiation regime, the incorporation of a polymeric plasticizer significantly enhances the efficiency of network development, leading to the most advanced crosslinked structure among the investigated systems.
EPR analysis of radical processes during thermoradiation vulcanization
The evolution of radical species during thermoradiation vulcanization of NBR-based systems was investigated by electron paramagnetic resonance (EPR) spectroscopy. This technique enables direct detection of paramagnetic intermediates generated during radiation–chemical crosslinking and provides mechanistic insight into radical initiation, recombination, and stabilization within the elastomeric network.
Figure 7 presents the EPR spectra of a thermoradiation-cured NBR formulation containing DMPM, polymeric plasticizer, sulfur, and ZnO, recorded (a) immediately after vulcanization and (b) after 48 h of storage under ambient conditions.
Immediately after vulcanization (Figure 7(a)), the EPR spectrum exhibits a well-resolved hyperfine structure (HFS), indicating a relatively high concentration of short-lived radical species generated during the active stage of network formation. The observed multiplet pattern, consisting of a doublet with additional fine splitting, is characteristic of carbon-centered radicals interacting with neighboring nuclei. This spectral feature reflects the involvement of low-molecular-weight components, including DMPM, sulfur, and polymeric plasticizer, in early radical-mediated processes and suggests that the radicals remain mobile and only weakly immobilized within the developing crosslinked network. 30
After 48 h of storage (Figure 7(b)), the EPR spectrum collapses into a weak, symmetric singlet, accompanied by a marked decrease in signal intensity. The disappearance of hyperfine interactions indicates extensive radical recombination and stabilization. The remaining signal is characterized by a g-factor of g = 2.0031 and a linewidth of ΔH = 0.44 mT, values typical of carbon-centered radicals trapped in polymer matrices. This spectral evolution demonstrates that radicals generated during thermoradiation curing are progressively immobilized as the network matures, leading to post-curing stabilization rather than sustained radical activity or degradation.
Reaction pathways involved in thermoradiation vulcanization of NBR systems
Based on EPR observations and established literature on sulfur vulcanization and radiation chemistry of elastomers, the thermoradiation curing of NBR proceeds through a sequence of radical-mediated reactions involving both polymer chains and sulfur-containing crosslinks. (1) Radical initiation under thermal and γ-irradiation exposure
Under combined thermal and γ-irradiation conditions, radical species are generated via homolytic cleavage of energetically labile bonds.
31
In sulfur-containing systems, polysulfidic bridges act as preferential radical sources: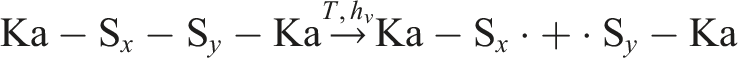
In parallel, γ-irradiation can induce radical formation directly along the unsaturated polymer backbone: (2) Hydrogen abstraction and formation of carbon-centered radicals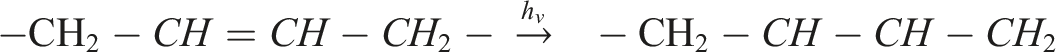
Sulfur-centered radicals readily abstract hydrogen atoms from neighboring polymer chains, leading to the formation of carbon-centered radicals: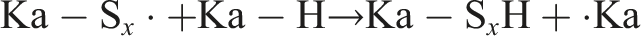
This step increases the concentration of carbon-centered radicals, which dominate the radical population detected by EPR during the early stages of curing. (3) Radical recombination and crosslink formation
The generated radicals undergo recombination reactions, resulting in the formation of new crosslinks with different chemical nature:
Carbon–carbon crosslinks (radiation-dominated): •Ka + •Ka → Ka–Ka.
Carbon–sulfur crosslinks (sulfur-assisted): •Ka + •Sy–Ka → Ka–Sy–Ka.
These reactions lead to the development of a hybrid crosslinked network composed of strong C–C linkages and shorter sulfur bridges. (4) Radical immobilization and network stabilization
As curing proceeds and the network density increases, radical mobility is progressively reduced. Residual radicals become immobilized within the crosslinked matrix, giving rise to weak, symmetric EPR singlets characteristic of trapped carbon-centered radicals: g = 2.0031, with ΔH = 0.44, was determined (Figure 7 (b)).
The decrease observed in EPR signal intensity suggests progressive radical recombination and stabilization of the crosslinked network rather than extensive ongoing degradation. This interpretation is consistent with the typical post-irradiation behavior of elastomeric systems reported in the literature. (5) Role of low-molecular-weight components
Low-molecular-weight additives, including the polymeric plasticizer and the maleimide co-agent (DMPM), enhance segmental mobility and facilitate radical diffusion, thereby promoting radical recombination over irreversible chain scission. Their presence shifts the reaction balance toward efficient network formation under thermoradiation conditions.
Mechanical and physico-mechanical performance of NBR vulcanizates cured by different mechanisms
Plastoelastic, technological, and physico-mechanical properties of NBR-based vulcanizates (SKN-40) obtained by different vulcanization routes and formulations.
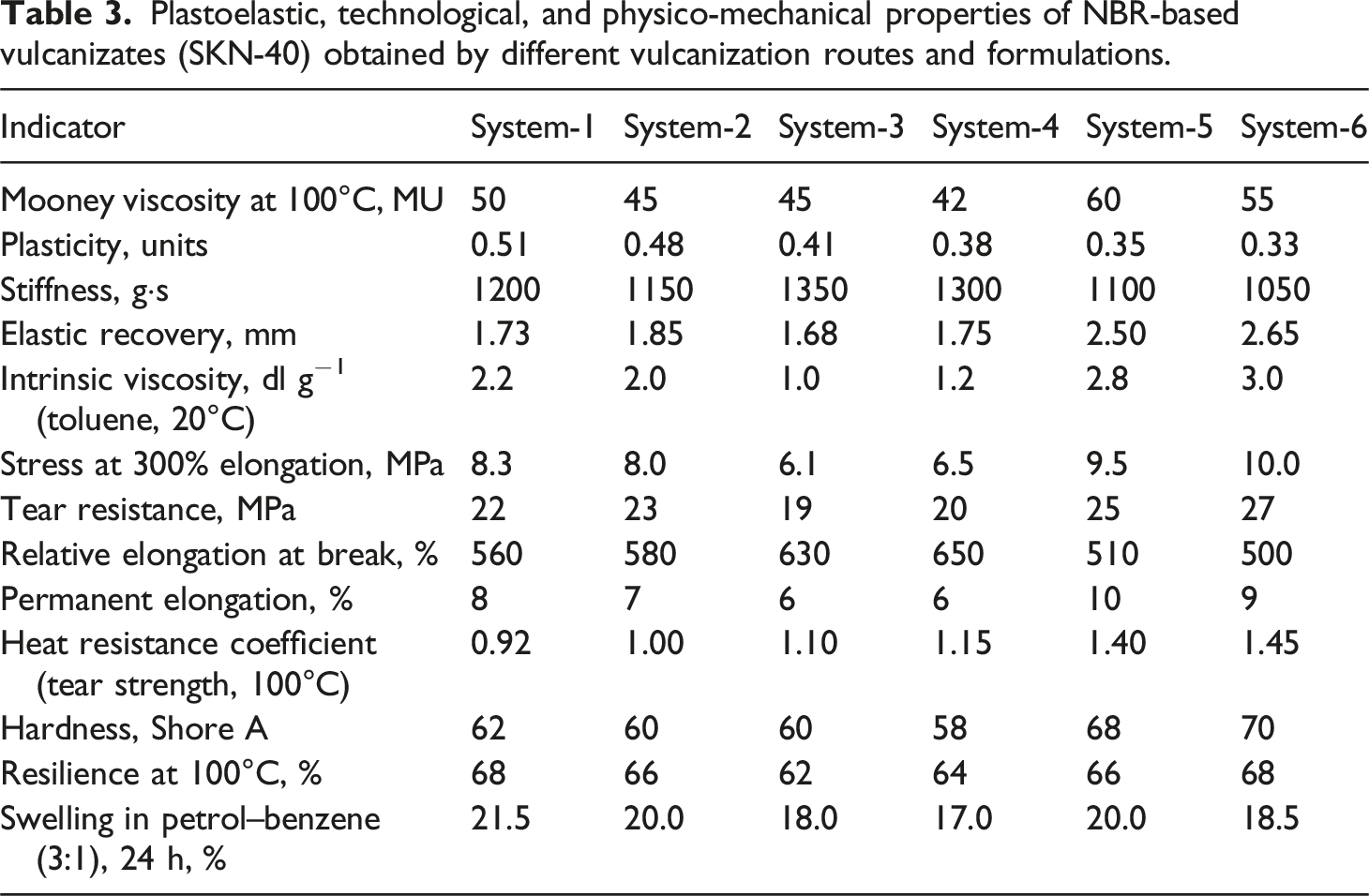
From a processing perspective, the Mooney viscosity data (Table 3) indicate that thermoradiation-cured systems exhibit the highest viscosity values. This behavior reflects the formation of a more developed and interconnected spatial network, which restricts chain mobility already at the processing stage. In contrast, radiation-cured samples show lower viscosity, suggesting that under irradiation alone, chain scission competes more strongly with crosslink formation, an effect widely reported for elastomers exposed to high-energy radiation in the absence of sulfur-mediated stabilization. 38
The evolution of plasticity and stiffness further clarifies these differences. A progressive decrease in plasticity from thermal to radiation and thermoradiation vulcanizates indicates increasing restriction of segmental mobility as crosslink density rises. Notably, thermoradiation samples combine relatively low plasticity with moderate stiffness, implying that the resulting network is dense yet structurally uniform, avoiding excessive rigidity. This balance is characteristic of hybrid networks in which strong carbon–carbon crosslinks provide load-bearing capacity, while shorter sulfur bridges contribute flexibility and stress redistribution. 39
Intrinsic viscosity measurements (Table 3) provide molecular-level evidence for these trends. The pronounced increase in intrinsic viscosity for thermoradiation vulcanizates indicates that crosslinking reactions dominate over degradation, leading to an effectively enlarged macromolecular structure.
By contrast, the low intrinsic viscosity of radiation-cured samples is consistent with partial backbone scission, a well-established consequence of γ-irradiation when radical recombination is insufficiently promoted.4,18 The presence of sulfur and DMPM in thermoradiation systems shifts this balance toward crosslink formation rather than molecular weight reduction.
Mechanical performance follows directly from these structural features. As shown in Table 3, thermoradiation vulcanizates exhibit the highest stress at 300% elongation and tear resistance, indicating a superior ability to sustain and redistribute applied loads. Radiation-cured samples retain high elongation at break, reflecting a more flexible but mechanically weaker network, whereas thermal vulcanizates show intermediate behavior dominated by sulfur crosslinks of varying length and thermal stability. These observations are consistent with the established understanding that C–C crosslinks impart higher strength and thermal robustness than polysulfidic C–Sx–C bonds.37,38
Thermal resistance characteristics provide particularly strong support for the hybrid curing concept. The heat resistance coefficient for tear strength increases markedly for thermoradiation vulcanizates (Table 3), reflecting the higher fraction of thermally stable carbon–carbon linkages supplemented by a limited number of short sulfur bridges. In contrast, the lower thermal stability of purely sulfur-cured samples is attributable to the susceptibility of polysulfidic bonds to thermal cleavage, while radiation-only networks lack the reinforcing contribution of sulfur-assisted crosslinks. 40
Solvent swelling behavior reinforces these conclusions. Radiation-cured vulcanizates display the lowest swelling degree in petrol–benzene mixtures, consistent with the impermeable nature of carbon–carbon crosslinks. 41 Thermoradiation samples show slightly higher swelling due to the presence of sulfur bonds, yet still significantly outperform thermally cured and control samples. This result highlights the compactness and efficiency of the hybrid network and confirms that the coexistence of C–C and C–Sx–C junctions does not compromise solvent resistance. 42
The polymeric plasticizer enhances segmental mobility and radical diffusion during irradiation, while sulfur contributes controlled secondary crosslinking during the thermal step. This synergistic interaction results in vulcanizates with a superior balance of processability, mechanical strength, thermal stability, and solvent resistance compared with materials cured by thermal or radiation routes alone.43–45 Such a combination of properties is difficult to achieve by conventional curing strategies and represents a meaningful advancement in the design of high-performance NBR materials.
Thermal stability and decomposition behavior of NBR vulcanizates under nitrogen atmosphere
Thermal degradation parameters of NBR (SKN-40) vulcanizates cured by different mechanisms, determined from TGA/DTG analysis under nitrogen atmosphere.
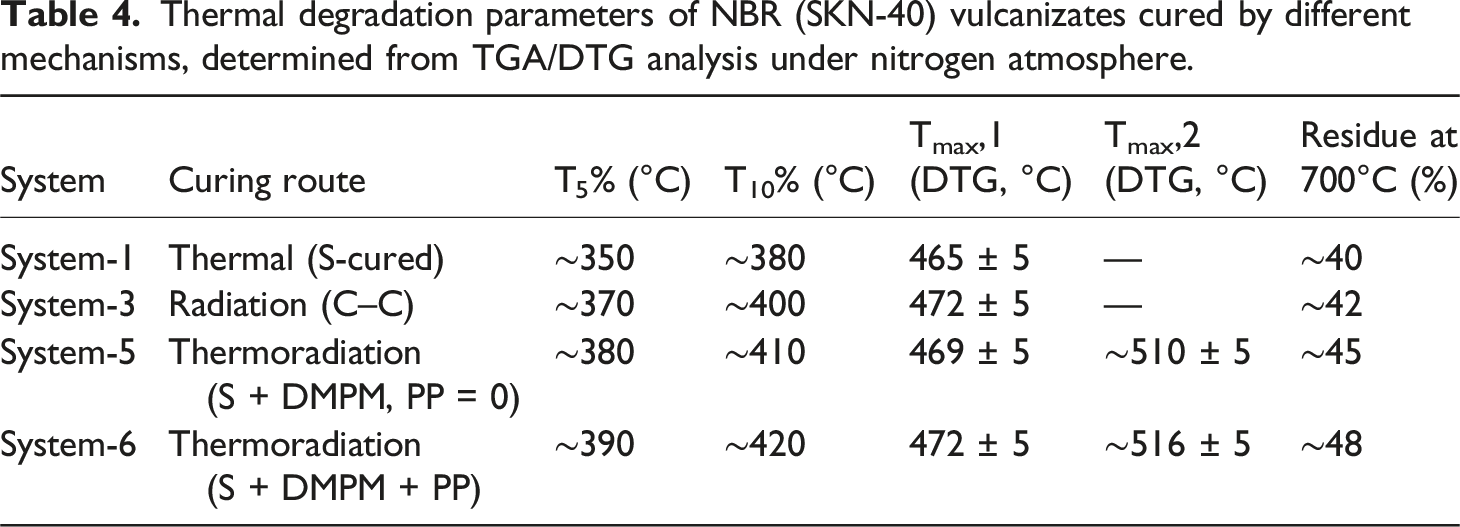
The thermally cured sulfur vulcanizate (System-1) exhibits the lowest thermal stability, with T5% ≈ 350°C and T10% ≈ 380°C. The DTG curve shows a single maximum at 465 ± 5°C, corresponding to the main-chain decomposition of the NBR backbone. The relatively early onset of mass loss is consistent with the presence of polysulfidic C–Sx–C crosslinks, which are thermally labile and prone to cleavage at elevated temperatures. The residue at 700°C is approximately 40%, indicating limited char formation.
Radiation-cured vulcanizates (System-3) demonstrate a noticeable improvement in thermal resistance. The onset temperatures increase to T5% ≈ 370°C and T10% ≈ 400°C, confirming that radiation-induced carbon–carbon crosslinks delay the initiation of thermal degradation. The DTG maximum shifts slightly to higher temperature (472 ± 5°C), while the residue increases marginally to ∼42%. These results indicate that C–C crosslinks provide enhanced thermal stability compared with sulfur bridges but still produce a relatively homogeneous degradation process characterized by a single DTG peak.
The results of the TG and DTG analysis of the NBR samples in inert nitrogen atmosphere are presented in Figure 8. TG and DTG curves of NBR (SKN-40) vulcanizates cured by different mechanisms under nitrogen atmosphere: (a) Thermal sulfur vulcanization (System-1); (b) Radiation vulcanization (System-3); (c) Thermoradiation vulcanization without polymeric plasticizer (System-5); (d) Thermoradiation vulcanization with polymeric plasticizer (System-6).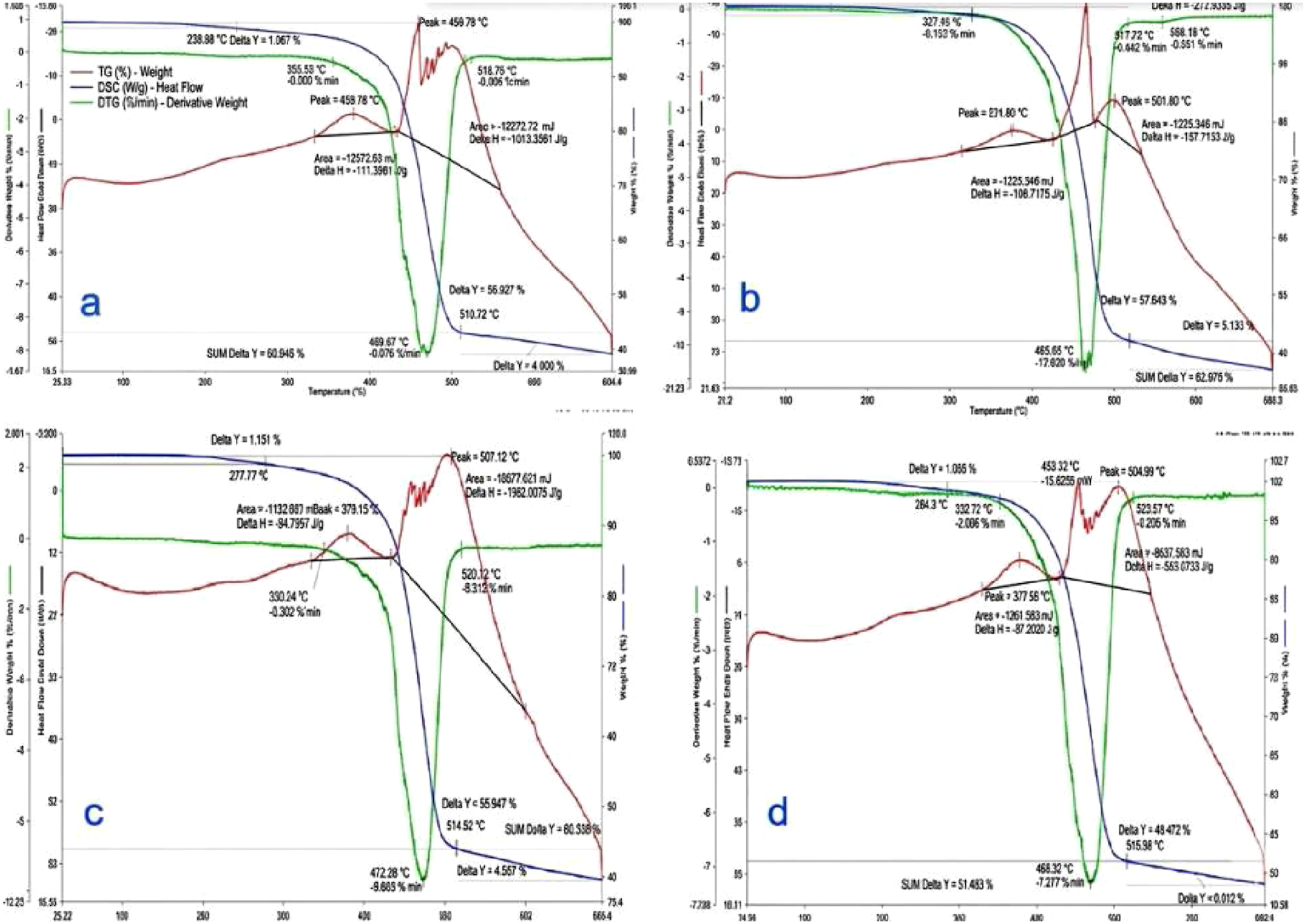
A further improvement is achieved in thermoradiation-cured systems. For System-5 (without polymeric plasticizer), T5% and T10% increase to approximately 380°C and 410°C, respectively. In contrast to thermal and radiation vulcanizates, the DTG curve exhibits two distinct maxima: the first at 469 ± 5°C, associated with backbone decomposition, and a second at ∼510 ± 5°C, attributed to the degradation of highly crosslinked and condensed network domains. The appearance of this second DTG peak provides direct evidence for a more complex and heterogeneous network formed under thermoradiation conditions. The residue content increases to ∼45%, reflecting restricted volatilization and enhanced char formation.
The highest thermal stability is observed for the polymer-plasticized thermoradiation vulcanizate (System-6). The onset temperatures reach T5% ≈ 390°C and T10% ≈ 420°C, representing the maximum values among all investigated systems. The DTG maxima are located at 472 ± 5°C and 516 ± 5°C, indicating further stabilization of both the primary polymer backbone and the crosslinked domains. The residue at 700°C increases to ∼48%, confirming the formation of the most compact and thermally resistant network. The presence of the polymeric plasticizer facilitates segmental mobility during curing, promoting more efficient radical recombination and crosslink consolidation rather than chain scission.
Overall, the TGA results demonstrate a clear correlation between curing route, crosslink chemistry, and thermal stability. Sulfur vulcanization produces networks with early degradation onset due to thermally unstable polysulfidic bonds. Radiation curing improves stability through the formation of carbon–carbon crosslinks but remains limited to a single-step degradation behavior. Thermoradiation curing generates a hybrid network containing both C–C and short C–S bonds, resulting in delayed degradation onset, multi-stage decomposition, and increased char residue. Among all systems, the polymer-plasticized thermoradiation vulcanizate exhibits the most advanced thermal performance, confirming the effectiveness of this curing strategy for high-temperature-resistant NBR applications.
Conclusion
This study demonstrates that the curing route and polymeric plasticizer content govern both the crosslinking mechanism and the resulting network structure of nitrile butadiene rubber (NBR). Sulfur vulcanization proceeds through the formation of polysulfidic and disulfidic bridges, which provide elasticity but undergo thermal rearrangement and scission, limiting thermal stability. Radiation vulcanization is controlled by radical generation along the NBR backbone, as confirmed by EPR analysis, which reveals the formation of carbon-centered macroradicals that recombine to form carbon–carbon crosslinks while competing with chain scission processes. The incorporation of a polymeric plasticizer increases segmental mobility and enhances radical diffusion, leading to faster radical recombination and lower steady-state radical concentrations observed by EPR. Under thermoradiation vulcanization, EPR data indicate a synergistic mechanism in which radiation-induced macroradicals coexist with sulfur-centered intermediates, enabling the simultaneous formation of carbon–carbon bonds and short sulfur bridges. This hybrid crosslinking mechanism results in the highest effective crosslink density, delayed thermal degradation (T5% ≈ 390°C, T10% ≈ 420°C), increased char residue (≈48%), and improved mechanical performance (stress at 300% elongation ≈10 MPa). Overall, polymeric plasticizer-assisted thermoradiation vulcanization provides a mechanistically well-defined and effective approach for controlling crosslink topology and enhancing the thermal and mechanical performance of NBR.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Azerbaijan Science Foundation - Grant No. AEF-MGC-2024-2(50)-16/12/4-M-12.