
Abstract
Chitosan, as a renewable biological macromolecule, has attracted increasing interest for membrane engineering owing to its abundant functional groups and strong affinity toward metal ions. In this study, waste-derived chitosan extracted from recycled oyster shells was blended with poly (vinyl alcohol) (PVA) and fabricated into electrospun nanofibers, which were subsequently grafted onto cellulose acetate reverse osmosis (CA-RO) membranes to form biopolymer composite membranes. The macromolecular structure, intermolecular interactions, and physicochemical properties of the resulting membranes were systematically investigated using SEM, XRD, and FTIR analyses. The electrospun CS/PVA fibers exhibited diameters ranging from 182 to 459 nm, with higher electrospinning voltages yielding thinner and more uniform fibers. Grafting of the CS/PVA fibers significantly enhanced membrane hydrophilicity, reducing the water contact angle from 68.21° to 51.2°, and improved mechanical performance, with tensile strength increasing from 86.17 MPa to 144.55 MPa. These improvements are primarily attributed to hydrogen bonding and macromolecular chain interactions between chitosan, PVA, and cellulose acetate. The functionalized composite membranes demonstrated high rejection efficiencies for Pb(II), Cu(II), and Cd(II), with minimal impact on water permeability. The CS/PVA/CA-RO composite membranes exhibited high heavy metal rejection efficiencies of 85.98%, 90.51%, and 95.31% for Pb(II), Cu(II), and Cd(II), respectively, while maintaining a water flux of 9.96 L·m−2·h−1 highlighting the effective integration of adsorption functionality with structural stability. This work presents a sustainable macromolecular engineering strategy for designing high-performance biopolymer composite membranes via electrospinning and surface grafting, offering promising potential for advanced separation applications.
Highlight
• Mussel shell-derived chitosan blended with PVA to fabricate nanofibers • CS/PVA fibers grafted onto CA-RO membranes, forming mechanically composites • Surface modification enhanced hydrophilicity and tensile strength • Efficient removal of Pb(II), Cu(II), and Cd(II) with minimal flux decline
Introduction
In recent years, biological macromolecules have emerged as key building blocks in advanced membrane materials due to their renewable origin, structural diversity, and rich chemical functionality. Among these materials, polysaccharide-based macromolecules such as chitosan offer unique advantages, including a high density of amino and hydroxyl groups, which enable strong intermolecular interactions, chemical tunability, and functional surface modification. 1 However, the practical application of chitosan-based membranes is often limited by poor mechanical strength and structural instability, necessitating macromolecular blending and hybridization strategies. Blending chitosan with synthetic polymers such as poly (vinyl alcohol) (PVA) represents an effective macromolecular engineering approach to tailor polymer chain interactions, enhance hydrogen bonding, and improve mechanical integrity while preserving functionality.2,3 Furthermore, electrospinning provides a versatile platform for controlling polymer chain orientation, fiber diameter, and surface area at the nanoscale, enabling precise structure property optimization. 4 When combined with surface grafting techniques, electrospun biopolymer fibers can be chemically anchored onto supporting substrates, forming hierarchical composite membranes with enhanced stability and multifunctional performance. Water contamination by heavy metals represents a critical environmental and public health issue worldwide, necessitating the development of efficient, sustainable, and cost-effective water treatment technologies. 5 Conventional reverse osmosis (RO) membranes have been widely employed for desalination and wastewater purification due to their high selectivity. However, these membranes often face limitations such as severe fouling, low adsorption capacity for heavy metals, limited surface functionalization, and high operational energy requirements. 6 Such challenges have motivated the exploration of hybrid and biopolymer-based membranes that integrate adsorption functionality with mechanical stability and processability. 7 Chitosan, a naturally occurring polysaccharide extracted from crustacean or oyster shells, has attracted increasing attention for membrane applications due to its biodegradability, biocompatibility, and strong affinity for heavy metal ions via its amino (-NH2) and hydroxyl (-OH) groups.8,9 Despite these advantages, pristine chitosan exhibits poor mechanical strength and limited water permeability, restricting its large-scale application in membrane technologies. 10 To overcome these limitations, chitosan can be blended with poly (vinyl alcohol) (PVA), a mechanically robust, water-soluble, and electrospinning polymer. The resulting CS/PVA blend benefits from enhanced structural integrity, improved hydrophilicity, and higher water flux.11,12 Electrospinning of CS/PVA fibers further creates highly porous networks with large surface area, providing abundant active sites for metal ion adsorption while facilitating water transport through the membrane.13–15 Cellulose acetate (CA) remains one of the most widely used polymers for RO membranes, offering excellent selectivity, chemical stability, affordability, and processability.16–19 However, pristine CA membranes suffer from limited antifouling resistance and weak removal efficiency for dissolved contaminants such as heavy metals.20–23 Grafting electrospun CS/PVA fibers onto CA-RO substrates represents a promising strategy to enhance surface functionality, adsorption efficiency, and overall membrane performance without compromising permeability.24,25 The novelty of this study lies in the integration of waste-derived materials and structured biopolymers into a sustainable hybrid membrane design. Chitosan is extracted from recycled oyster shells, valorizing a common bio-waste and contributing to circular economy principles. The CS/PVA fibers are electrospun and grafted onto CA-RO membranes, forming a composite platform that combines the adsorptive capacity of biopolymers with the mechanical strength, selectivity, and durability of commercial substrates. This study builds upon the convergence of three key trends in membrane research: (i) the utilization of renewable and recycled raw materials, (ii) structured membrane fabrication through electrospinning, and (iii) functional surface grafting to enhance adsorption and separation performance. The CS/PVA-CA composite membranes are anticipated to provide a sustainable and high-performance solution for industrial wastewater treatment, addressing both environmental and technological challenges simultaneously.
Materials and methods
Materials
Recycled oyster shells were initially gathered from a nearby fish market and prepared for use in the study. Acetic acid (glacial, 100%, pro-analyzed grade), sodium hydroxide, poly (vinyl alcohol) (PVA), and potassium persulfate were all supplied by El Gomhouria Company for Trading. Bovine serum albumin (BSA, ≥98%) was used as a model protein foulant. Acetone with purity greater than 99% was obtained from Chemika (India). Cellulose acetate (CA), with an approximate molecular weight of 100,000 g/mol, was purchased from Aldrich (USA). Standard stock solutions of Pb(II), Cu(II), and Cd(II), each at a concentration of 1000 mg/L, were utilized for evaluating the heavy-metal removal performance. Additionally, 1,4-dioxane was procured from Panreac Química S.A. (Spain).
Chitosan extraction from recycled oyster shells
Chitosan was produced from recycled oyster shells through a sequence of controlled physical and chemical treatments. The shells were first washed thoroughly with distilled water to remove residual organic debris, salts, and surface impurities. After cleaning, they were dried in a hot-air oven at 70°C for about 7 hours to eliminate moisture and allow efficient milling. The dried shells were then ground into a fine powder and subjected to a demineralization step, during which the powder was immersed in 1.0 M hydrochloric acid at room temperature (25°C) for 2 hours to dissolve calcium carbonate and other inorganic components. Following the acid treatment, the solid fraction was repeatedly rinsed with distilled water until a neutral pH was achieved. A subsequent deproteinization stage was carried out by treating the demineralized material with 1.5 M sodium hydroxide at 70°C for 2 hours to remove remaining protein residues. The reaction produced a brown suspension that was filtered to isolate the solid chitin phase. The obtained chitin was washed several times with distilled water to remove any traces of alkali, producing a pale-white suspension as shown in Figure 1. The purified chitin was finally dried in an oven at 60°C to yield a fine white powder. To maintain uniformity among samples, all collected oyster shells were pooled and processed under the same experimental conditions. Multiple washing and drying steps effectively removed soluble impurities, reduced microbial contamination, and improved the overall purity of the extracted chitin, which was later converted into chitosan through standard deacetylation. Preparation of chitosan from recycled oyster shells: (a) drying and grinding, (b) crushing into smaller pieces, (c) chemical treatment to remove impurities, (d) purified chitosan powder.
Chitosan/Poly (Vinyl Alcohol) Blended Film
A chitosan solution was prepared by dissolving 4 g of chitosan in 400 mL of 2% (v/v) acetic acid. The mixture was stirred continuously and maintained at approximately 60°C for 10 h to ensure complete dissolution. The resulting viscous solution was then filtered to remove any undissolved residues or impurities and subsequently left undisturbed at room temperature for about 2 h to allow trapped air bubbles to dissipate. Separately, a poly (vinyl alcohol) (PVA) solution was obtained by dissolving 4 g of PVA in 400 mL of deionized water, which had been preheated to about 85°C. The mixture was stirred until complete dissolution, yielding a clear homogeneous solution. Separately, a poly (vinyl alcohol) (PVA) solution was prepared by dissolving 4 g of PVA in 400 mL of deionized water preheated to approximately 85°C. The mixture was stirred until complete dissolution, yielding a clear and homogeneous solution. To prepare the blended chitosan/PVA (CS/PVA) solution, the two polymer solutions were mixed at a volumetric ratio of 60:40 (CS: PVA). This ratio was selected to achieve a suitable balance between mechanical strength and flexibility, where chitosan contributes film-forming ability and bioactivity, while PVA enhances elasticity and flexibility. The chosen ratio was found to provide homogeneous films with satisfactory tensile properties, as supported by previous studies.18–20 The PVA solution was gradually added to the chitosan solution under constant magnetic stirring at approximately 90°C for 30 min to ensure uniform mixing and effective molecular interaction between the two polymers. The resulting blend was then ready for casting into films for subsequent characterization.
Preparation of Chitosan/Poly (Vinyl Alcohol) Fibers
Electrospun chitosan/PVA fibers were fabricated using a commercial electrospinning setup, comprising a syringe pump, stainless-steel needle spinneret, high-voltage DC supply, and a grounded collector. The blended polymer solution was loaded into a 10 mL plastic syringe and delivered at a constant flow rate of approximately 0.8 mL h−1, selected based on preliminary trials to ensure stable jet formation and avoid bead formation. A high-voltage DC supply was applied in the range of 22–28 kV to investigate the influence of voltage on fiber morphology and uniformity. The tip-to-collector distance was fixed at 14 cm throughout all experiments to minimize variability and maintain a stable electric field. Accordingly, the electrospinning parameters were kept constant to isolate the effect of material composition on fiber morphology. The collector consisted of a grounded flat copper plate covered with aluminum foil for uniform fiber deposition. The positive terminal of the power supply was connected to the needle, and the negative terminal to the collector. Electrospinning was conducted at room temperature (∼25°C) and ambient humidity (∼50% RH). After fabrication, the fibrous mats were carefully removed and stored in a desiccator containing silica gel to remove residual moisture and preserve their structural integrity prior to characterization.
Preparation of Cellulose Acetate (CA-RO) Membrane
The cellulose acetate reverse osmosis (CA-RO) membranes were produced using a solvent–nonsolvent casting approach. To prepare the polymer dope, 8.45 g of cellulose acetate was dissolved in a mixed solvent system composed of dioxane (27.6 g), acetone (10.6 g), and acetic acid (5.1 g), while methanol (8.4 g) was incorporated as the nonsolvent component. The mixture was stirred continuously at room temperature for 24 hours to ensure complete polymer dissolution. To remove air bubbles, the resulting homogeneous solution was placed in an ultrasonic bath and sonicated for 30 minutes. The degassed dope was then spread on a clean glass plate using an automatic film applicator (Zehntner 2300, Switzerland). A casting thickness of 250 μm and a speed of 10 mm/s were maintained at ambient conditions. After allowing the film to evaporate for approximately 60 seconds, it was immediately transferred into an ice-cold (<4°C) deionized water coagulation bath for 15 minutes to induce phase inversion and membrane formation, as illustrated in Figure 2. The newly formed membranes were kept submerged in the cold bath for an additional 2 hours to release internal stresses and reduce capillary forces. Following coagulation, the membranes were washed thoroughly with distilled water to remove residual solvents. Subsequent post-treatment involved immersing the membranes in a hot-water bath at 80°C for 10 minutes, followed by soaking them in deionized water for 24 hours. Finally, the membranes were air-dried for another 24 hours prior to being used in further modification or characterization steps. Fabrication of the CA-RO membrane via casting: (a) degassing the polymer solution, (b) spreading on a glass plate, (c) applying uniform thickness with a film applicator, (d) wet membrane formation, (e) immersion in coagulation bath, (f) final membrane after phase inversion.
Grafting of CS/PVA Fibers onto CA Membranes
The composite cellulose acetate (CA-RO) membrane incorporated with chitosan/poly (vinyl alcohol) (CS/PVA) fibers was prepared using a surface grafting technique. Initially, the CA-RO membrane was mildly treated with 0.04% sodium hydroxide solution for 5 minutes to partially deacetylate the surface and expose reactive hydroxyl groups. The thickness of the electrospun CS/PVA fiber mat deposited onto the CA membrane was estimated from SEM cross-sectional images and found to be approximately 10–25 µm, depending on deposition density. After thorough rinsing with deionized water, the membrane surface was activated by the drop-wise addition of 1.5 wt% potassium persulfate (K2S2O8) for 10 minutes, generating free radicals that facilitated graft formation. Pre-prepared electrospun CS/PVA fibers were then carefully deposited on the activated CA surface and gently pressed to ensure uniform contact. The grafting reaction was allowed to proceed at room temperature for 10 minutes, enabling the fibers to chemically bond with the CA layer Figure 3. After completion, the composite membranes were repeatedly washed with deionized water to remove any unreacted residues, followed by thermal curing at 75°C for 30 minutes. The resulting CS/PVA/CA-RO fiber composite membranes were finally soaked in deionized water for 24 hours and air-dried before characterization. Fabrication of CS/PVA-CA composite membrane via electrospinning and grafting, showing heavy metal rejection and water permeation.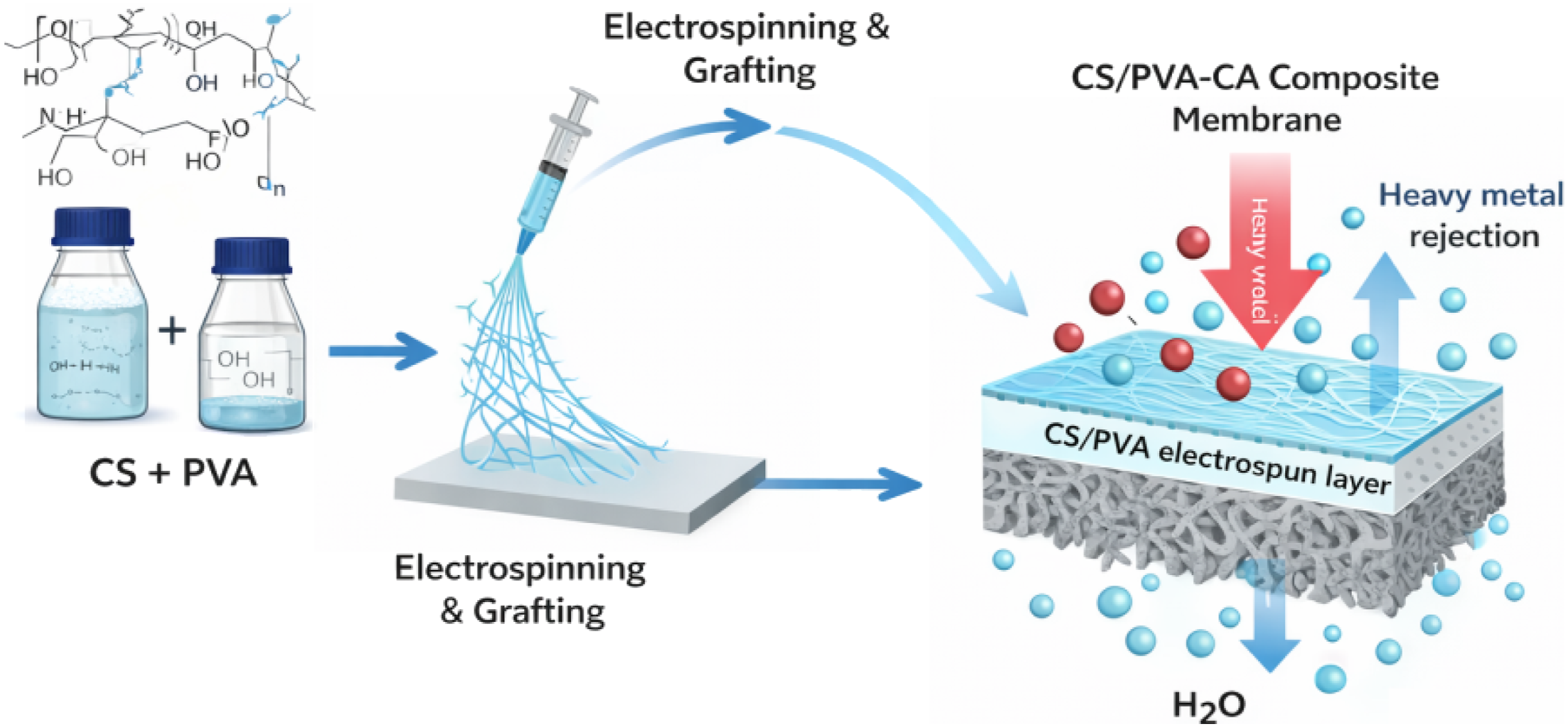
Antifouling activity
Bovine serum albumin (BSA) was selected as a model protein to evaluate membrane fouling behavior. Protein concentration was determined using UV–Vis spectrophotometry at a wavelength of 280 nm. A calibration curve was constructed using standard BSA solutions with concentrations ranging from 0 to 1000 mg/L, showing good linearity (R2 > 0.99). To evaluate the membranes’ susceptibility to protein fouling, square membrane pieces (2.5 × 2.5 cm) were placed in 10 mL of a protein solution and incubated in a shaking water bath at 25°C for 24 hours. The protein concentrations prior to contact with the membrane (C0) and after incubation (Ca) were quantified using a pre-established calibration curve. The relative protein adsorption (RP%) was then determined using equation (1):

Characterization
The chemical composition of the prepared membranes was analyzed using Fourier transform infrared spectroscopy (FTIR) on a Spectrum BX-11 spectrometer (Perkin Elmer, LX 18-5255). For cellulose acetate (CA) membranes, spectra were recorded over the range of 4000–400 cm−1. CA powders were mixed with potassium bromide (KBr) at a 10:1 ratio and pressed into transparent pellets using a hydraulic press to allow efficient IR beam transmission, whereas membrane samples were analyzed directly in their thin-film form. The surface morphology and cross-sectional structures of the CA membranes were examined by scanning electron microscopy (SEM, JEOL XL-30). Samples were cryo-fractured in liquid nitrogen to produce smooth cross sections, mounted on brass stubs using double-sided adhesive tape, and sputter-coated with a thin layer of gold to enhance image quality. Membrane surface wettability was assessed using a contact angle goniometer (Rame-Hart, USA). A 2 µL drop of distilled water was placed on a 3 × 2 cm section of the membrane surface using a Hamilton micro-syringe, and measurements were recorded at five different points within 30 seconds; the average value was reported. Heavy-metal adsorption capacities were determined using inductively coupled plasma spectroscopy (ICP, Shimadzu 7000, Japan). The crystalline phases of the samples were identified by X-ray diffraction (XRD) using a Shimadzu diffractometer. Pure-water permeation through the CA sublayers was measured at 25°C using a flow-through cell. The filtration performance of the composite membranes was evaluated by determining the flux and rejection for Cu, Cd, and Pb solutions (50 mg/L) under a pressure of 0.5 MPa. The permeate flux (F) was calculated according to equation (2):


Results and discussion
Structure investigation
Fourier-transform infrared (FTIR) spectroscopy was employed to investigate the structural features of pure polyvinyl alcohol (PVA) and chitosan (CS), as shown in Figure 4. For PVA, the broad absorption band centered around 3236 cm−1 is attributed to the O–H stretching vibrations of hydroxyl groups, indicating the presence of extensive hydrogen bonding within the polymer chains.
26
The peak observed around 2920 cm−1 is attributed to the C–H stretching vibrations of alkyl groups, confirming the structural integrity of the PVA backbone. For chitosan (CS), a broad absorption band within 3400–3200 cm−1 corresponds to overlapping O–H and N–H stretching vibrations, reflecting strong intra- and intermolecular hydrogen bonding, which is characteristic of its polyhydroxylated and amino-functional structure. The absorption near 2900 cm−1 is associated with aliphatic C–H stretching, while the sharp band at approximately 1650 cm−1 corresponds to the amide I (C = O stretching) vibration.
27
Furthermore, the distinctive peaks between 1220 and 1004 cm−1 are indicative of C–O–C stretching vibrations of the glycosidic linkages within the chitosan backbone. These spectral observations confirm that the key functional groups of the polymers were preserved, demonstrating that their chemical structures remained intact during preparation and ensuring their suitability for further modification or composite formation. FTIR spectra absorption bands of chitosan (CS) and polyvinyl alcohol (PVA).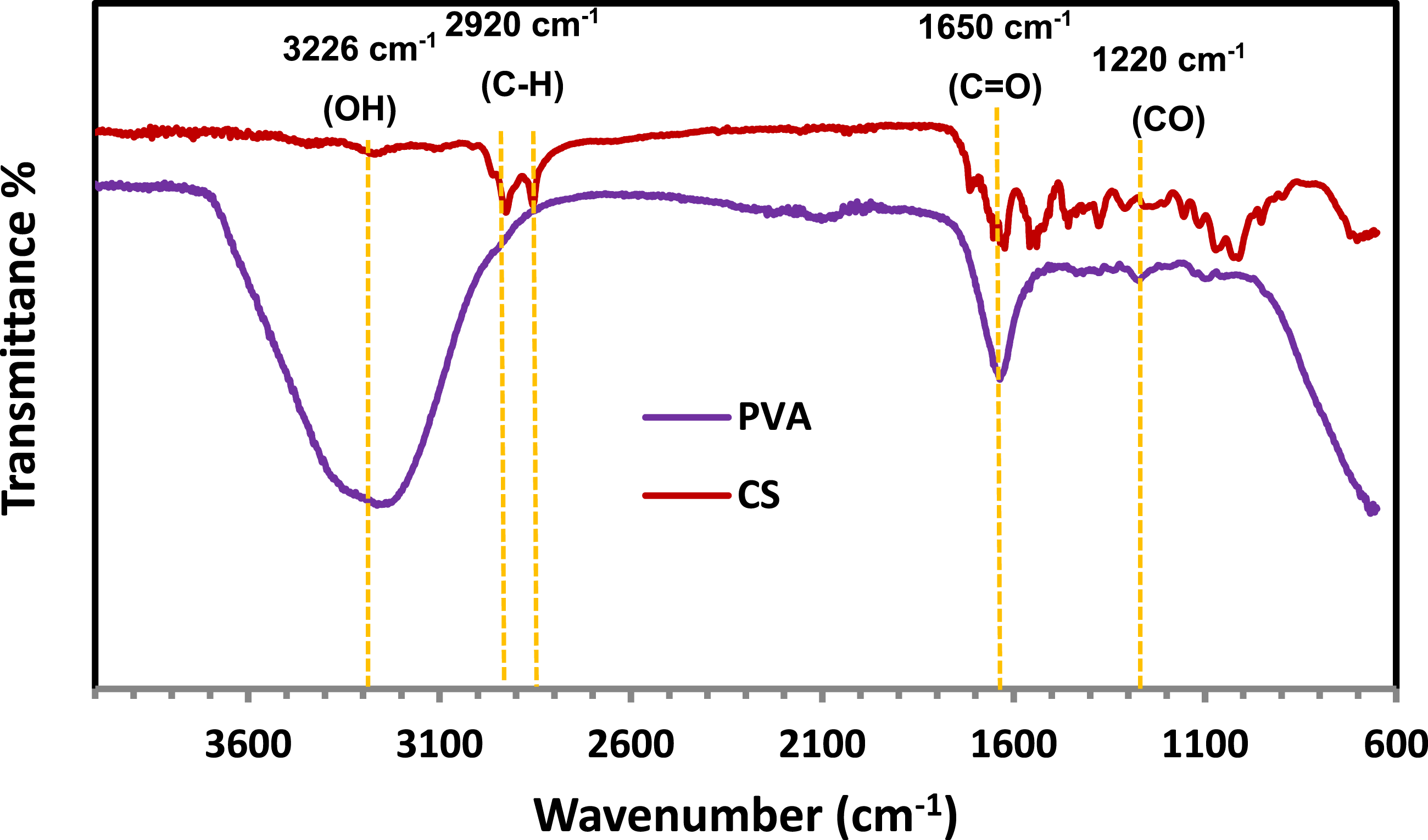
The degree of deacetylation (DD) of the extracted chitosan was estimated using FTIR spectroscopy based on the absorbance ratio of the amide I band at 1655 cm−1 to the hydroxyl band at 3450 cm−1, following reported methods in the literature. The calculated DD value was approximately (70-80%), which is within the typical range reported for chitosan derived from oyster shells. This DD level indicates a sufficient content of free amino groups capable of interacting with metal ions through chelation and electrostatic attraction.
FTIR spectroscopy was utilized to evaluate the structural integrity of the cellulose acetate (CA) membrane and to confirm the successful incorporation of chitosan/PVA into the CA matrix. As shown in Figure 5, the spectra display characteristic absorption bands corresponding to the functional groups of both CA and chitosan/PVA. In the spectrum of the pristine CA membrane, a prominent absorption at approximately 1730 cm−1 is assigned to the C = O stretching vibration of ester groups, verifying the preserved acetylated structure of cellulose acetate.
28
The bands observed in the 1230–1260 cm−1 range are attributed to C–O stretching vibrations, while the peak around 1030 cm−1 corresponds to C–O–C ether linkages, confirming the structural stability of the CA backbone. A medium-intensity band near 1370 cm−1 is related to C–H bending vibrations. Upon incorporation of chitosan/PVA, significant spectral changes were observed. A broad band appeared between 3400 and 3200 cm−1, corresponding to overlapping O–H and N–H stretching vibrations, indicating the introduction of hydroxyl and amino functionalities from chitosan and PVA. Additionally, new peaks at approximately 1650 cm−1 (amide I) and 1550 cm−1 (amide II) confirm the presence of chitosan, which contains residual acetylated amino groups. Minor shifts and broadening in the O–H/N–H and C = O regions suggest the formation of hydrogen bonds and electrostatic interactions between the CA matrix and the incorporated chitosan/PVA chains.
29
These observations demonstrate good chemical compatibility and homogeneous dispersion of the polymers within the CA network. Furthermore, the reduced intensity of the O–H stretching band (3200–3500 cm−1) relative to native CA supports the successful modification and blending process, leading to improved interfacial adhesion and enhanced structural stability of the composite membrane.
30
FTIR spectra of CA membrane and CS/PVA/CA-RO composite highlighting functional group modifications.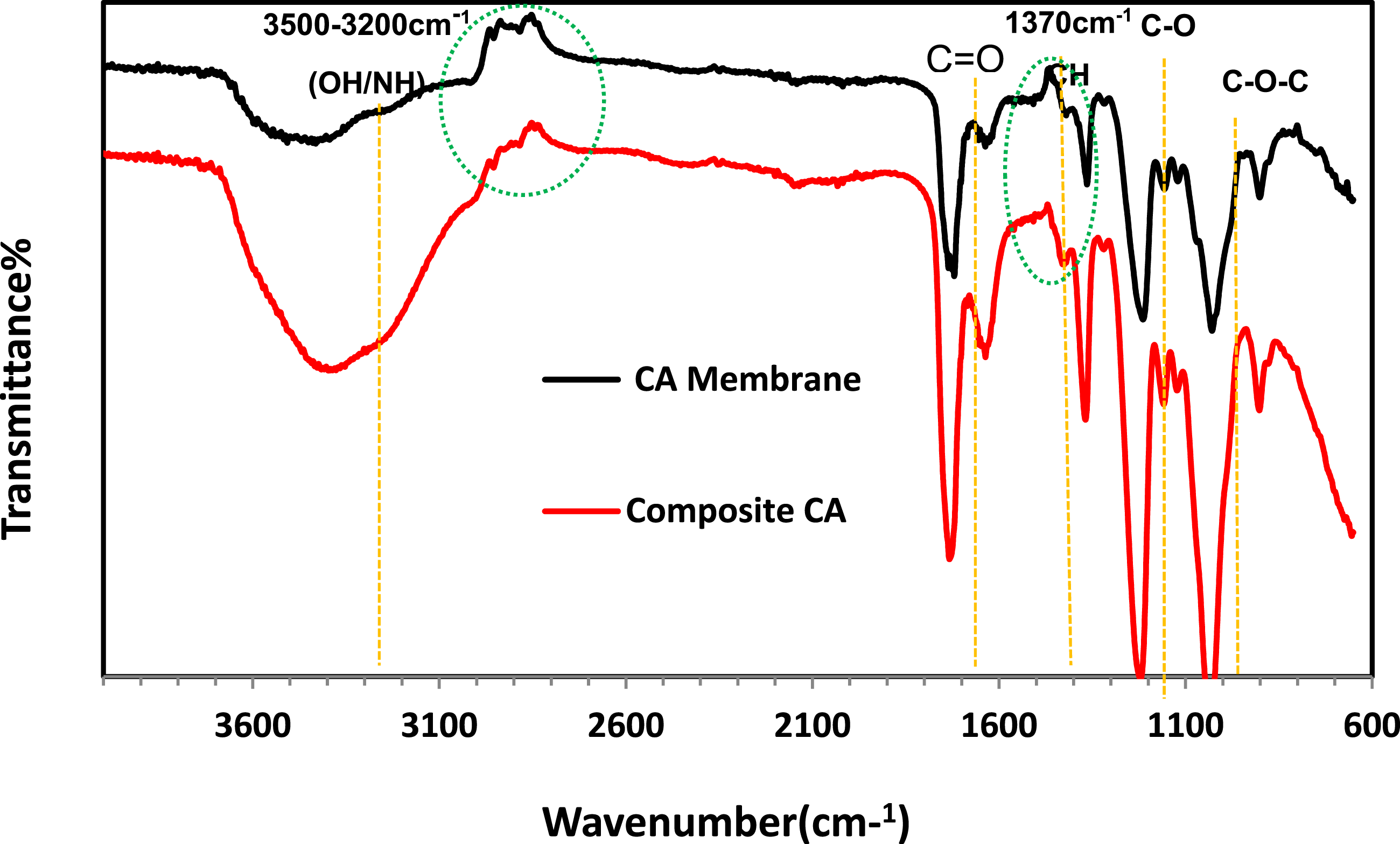
X-ray diffraction
Previous studies have shown that the chitosan (CS) film exhibits two characteristic crystalline peaks at 2θ = 15° and 20°. As illustrated in Figure 6(a), these peaks correspond to the (110) and (220) reflection planes, respectively. The rigid crystalline structure of CS is mainly attributed to intra-molecular and intermolecular hydrogen bonding, which defines the average intermolecular distances within the crystalline regions. Polyvinyl alcohol (PVA) can form strong intra-molecular and intermolecular hydrogen bonds through its OH groups, allowing sufficient interaction with the polymer backbone. Figure 6(b) shows the XRD patterns of pure CS and PVA films. The semi-crystalline nature of PVA is evident from a prominent peak at 2θ = 18°, while the amorphous regions are indicated by a broad peak around 2θ = 30°. When CS and PVA are combined, the intensity of the diffraction peaks increases and slightly broadens, suggesting partial disruption of hydrogen bonds due to the predominance of the amorphous structure in the blend. This behavior indicates that polymer blending is an effective method to reduce the crystalline regions of PVA.
31
The XRD patterns of the composite CS/PVA/CA-RO membranes display more complex features, as illustrated in Figure 6(c). Two broad halos were detected: one centered at 2θ ≈ 13° and a second, broader and more intense halo at 2θ ≈ 32°. These halos suggest the presence of multiple amorphous components with different short-range orders or inter-chain spacings.
32
Overall, the XRD analysis confirms that the incorporation of CS/PVA does not induce significant crystallinity in the composite membranes. The crystallinity of chitosan was evaluated qualitatively using XRD analysis, confirming its semi-crystalline nature.33,34 These characteristics confirm the suitability of the extracted chitosan for membrane fabrication and surface functionalization applications. As a result, the CS/PVA/CA-RO membranes retain a predominantly amorphous structure, which is expected to enhance flexibility and permeability critical properties for efficient membrane filtration applications. XRD patterns of: (a) chitosan CS, (b) CS/PVA and (c) CS/PVA/CA-RO membranes.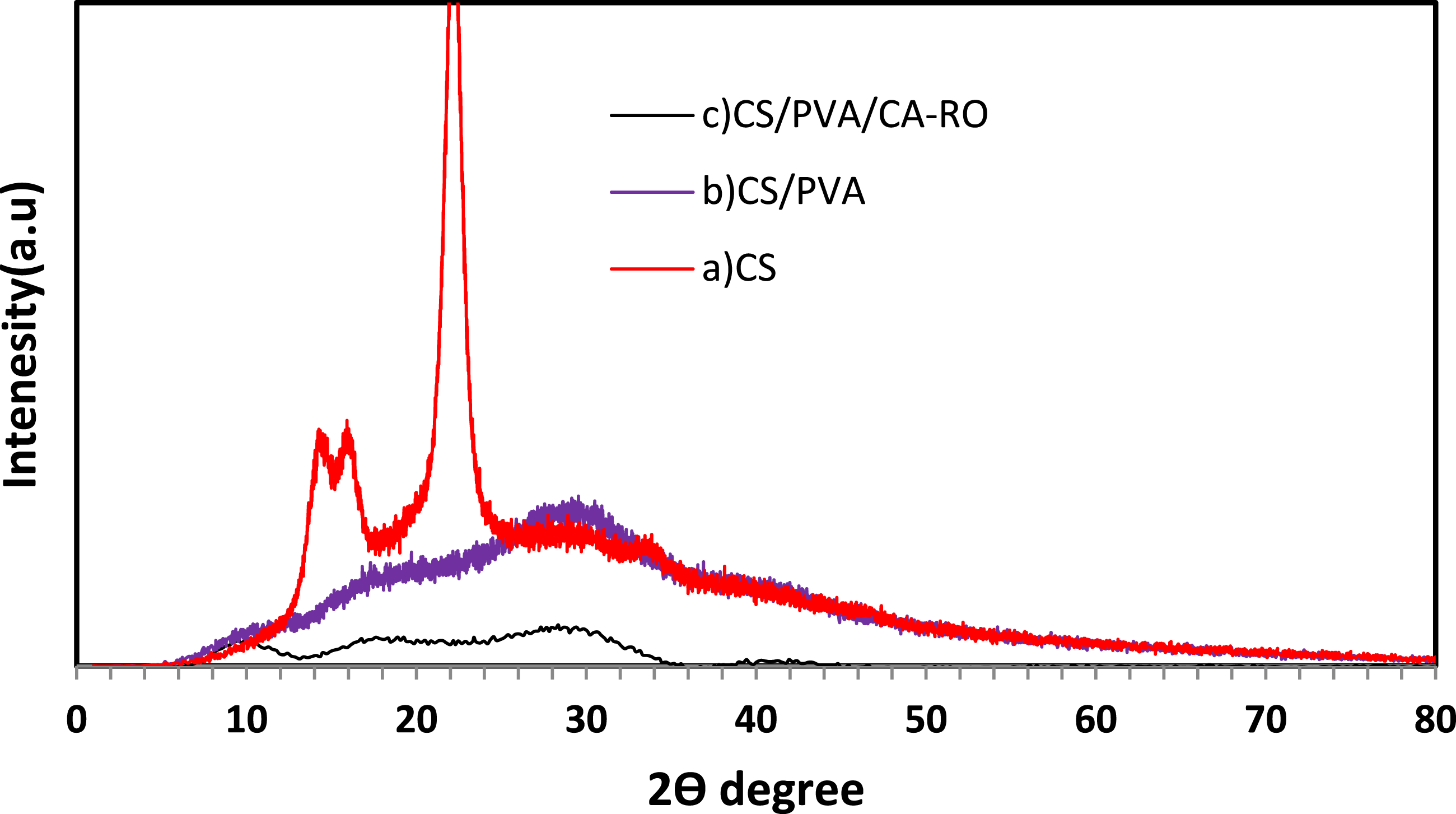
Morphological
Figure 7 illustrates the influence of applied voltage on the morphology and fiber diameter distribution of electrospun CS/PVA fibers, while all other electrospinning parameters were kept constant. The SEM micrographs and corresponding histograms clearly demonstrate that the applied voltage plays a decisive role in controlling fiber diameter and uniformity. At the higher applied voltage of 28 kV (Figure 7(a)), enhanced electrostatic stretching results in finer and more uniform fibers. A total of 44 fibers were randomly measured from SEM images using ImageJ software. The fibers exhibit an average diameter of 159 ± 37 nm, with diameters mainly distributed in the range of 100–263 nm. The relatively compact histogram indicates stable electrospinning conditions and effective elongation of the polymer jet under strong electrostatic forces. In contrast, fibers electrospun at the lower applied voltage of 22 kV (Figure 7(b)) show a noticeably broader diameter distribution. Analysis of 40 fibers reveals an average diameter of 236 ± 66 nm, with fiber diameters extending from approximately 170 to 460 nm. The appearance of thicker fibers at lower voltage can be attributed to reduced electrostatic drawing forces, which limit jet stretching and promote jet instability and partial fiber fusion, leading to less uniform fiber morphology. Although solution viscosity generally influences fiber diameter, the present results clearly demonstrate that electrostatic forces dominate over viscosity in the electrospinning of CS/PVA fibers. Increasing the applied voltage significantly enhances electrostatic stretching, resulting in finer and more uniform fibers, whereas lower voltage leads to thicker fibers with broader size distributions. These findings confirm that the applied voltage is a critical parameter for tailoring fiber morphology in electrospun CS/PVA membranes. (a) Shows the SEM micrograph and diameter histogram of fibers electrospun at 28 kV (collector distance 14 cm, flow rate 0.8 mL h−1), while Figure 7(b) displays fibers at 22 kV under the same conditions.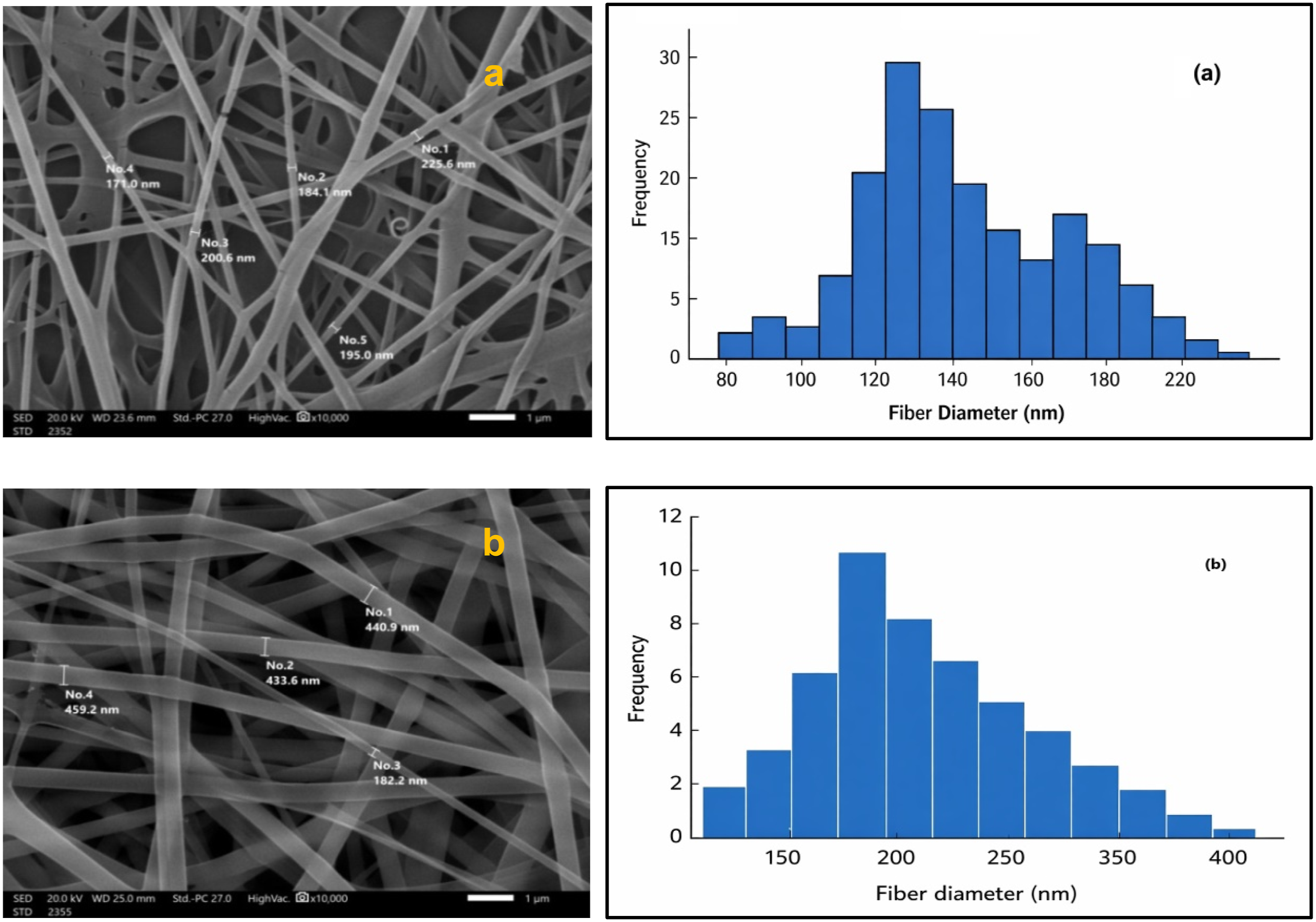
Figure 8 presents the scanning electron microscopy (SEM) images of the prepared membranes, illustrating the morphological differences between the pristine cellulose acetate reverse osmosis membrane (CA-RO) and the modified CS/PVA/CA-RO membrane. The images are presented in three distinct sections, representing the surface, bottom, and cross-sectional morphologies of each membrane. These micrographs provide valuable insight into the influence of chitosan (CS) and poly (vinyl alcohol) (PVA) grafting on the overall membrane morphology, porosity, and structural uniformity.
35
A clear distinction can be observed between the two membranes in terms of surface roughness, pore distribution, and the development of the characteristic finger-like structure in the cross-section. At the surface, the pristine CA-RO membrane (a) exhibits a relatively smooth and dense morphology with fine pores uniformly distributed, which is typical for cellulose acetate membranes. In contrast, the modified CS/PVA/CA-RO membrane (b) shows a noticeably rougher surface with interconnected fibrous and granular structures. This morphological transformation confirms the successful grafting of chitosan (CS) and poly (vinyl alcohol) (PVA) onto the CA matrix, which enhances surface hydrophilicity and improves antifouling properties. At the bottom, the pristine membrane displays a compact and less porous layer, while the modified membrane reveals an increased number of pores and surface roughness.
36
This indicates that the incorporation of CS and PVA altered the phase inversion dynamics, promoting better solvent-nonsolvent exchange during membrane formation and consequently enhancing water permeability. In the cross-section, both membranes exhibit a typical asymmetric structure; however, the pristine CA-RO membrane Figure 8(a) presents elongated finger-like pores extending from the dense top layer toward the sub-layer. The modified CS/PVA/CA-RO membrane Figure 8 (b) shows a more open and interconnected finger-like structure, suggesting improved pore connectivity and a more uniform internal morphology. This structural evolution confirms that the grafting process enhanced the interaction among polymer chains, resulting in a membrane with improved mechanical integrity, higher porosity, and superior hydrophilicity. Presents the SEM micrographs of (a) the unmodified CA-RO membrane and (b) the CS/PVA/CA-RO composite membrane, highlighting the surface, bottom, and cross-sectional.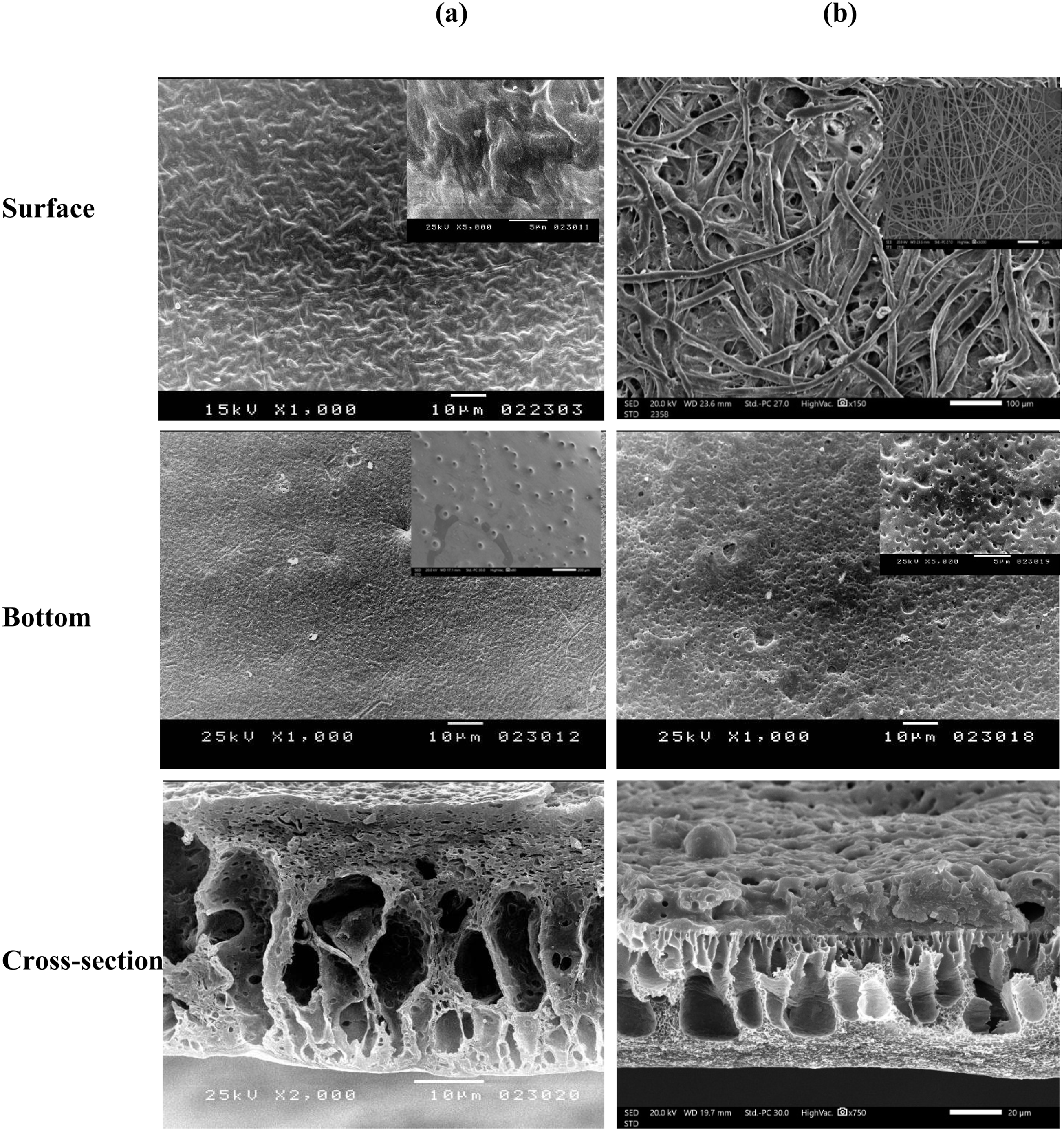
Separation of heavy metals
Heavy metal ion rejection performance and water flux of pristine and modified cellulose acetate (CA) membranes.
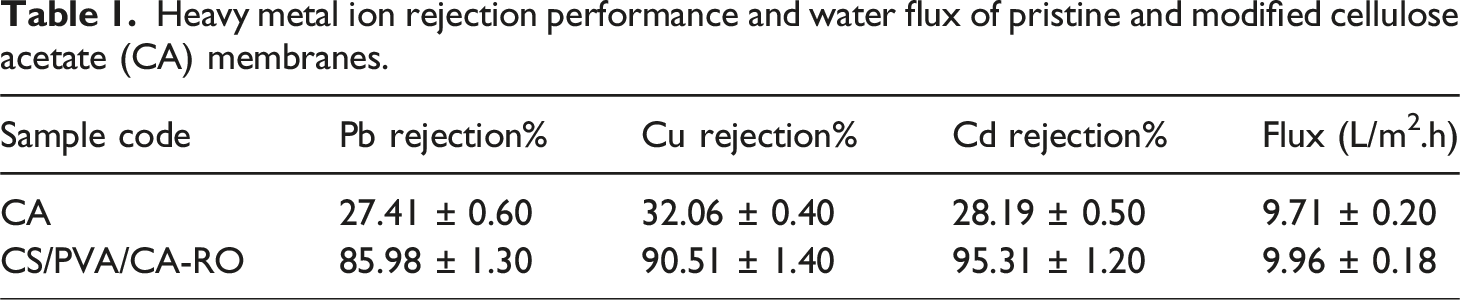
Similar trends have been reported for other particle- or polymer-modified CA membranes, where improved rejection occurs at the expense of a marginal flux reduction due to the formation of a denser selective layer. Overall, the results demonstrate that the CS/PVA/CA-RO membrane exhibits superior performance in heavy metal separation, achieving higher rejection efficiencies while maintaining reasonable permeability. The synergistic combination of CS and PVA effectively enhances the adsorption capability, surface polarity, and mechanical integrity of the CA matrix, making it a promising candidate for efficient removal of toxic metal ions from industrial wastewater. The metal ion removal mechanism involves a synergistic combination of size exclusion, surface adsorption, and chelation. The amino (–NH2) and hydroxyl (–OH) groups of chitosan act as primary binding sites, forming coordination complexes with Pb2+, Cu2+, and Cd2+ ions. Meanwhile, the hydrophilic PVA matrix enhances water transport and maintains structural stability, resulting in high rejection efficiency with minimal permeability loss.
Contact angle
The pristine cellulose acetate (CA) membrane showed a contact angle of 68.21° ± 1.2, reflecting its moderately hydrophobic nature, which can be attributed to the presence of acetyl functional groups along the polymer backbone. After grafting with chitosan (CS) and poly (vinyl alcohol) (PVA), the contact angle markedly decreased to 51.2° ± 1.0, confirming a substantial enhancement in surface hydrophilicity. This improvement can be attributed to the introduction of hydrophilic hydroxyl (–OH) and amine (–NH2) groups from CS and PVA, which increase the membrane’s affinity toward water molecules through hydrogen bonding and electrostatic interactions. The decrease in contact angle reflects a lower interfacial energy between the membrane and water, thereby facilitating improved wettability and faster water transport across the CS/PVA/CA-RO membrane surface. 40 Enhanced hydrophilicity also plays a crucial role in mitigating hydrophobic fouling by minimizing the adhesion of organic macromolecules and microbial species. Furthermore, the grafting of CS/PVA introduces additional surface roughness and polar functional sites, which collectively enhance water spreading behavior and contribute to the overall antifouling and antibacterial performance of the membrane. 41 These findings clearly demonstrate that the modification of CA with CS and PVA not only alters surface chemistry but also strengthens the functional synergy between hydrophilicity, permeability, and selectivity, leading to a more efficient and durable membrane suitable for wastewater treatment applications.
Mechanical properties
Mechanical properties of the prepared membranes.

Antifouling performance
To directly evaluate antifouling properties, a static protein adsorption (BSA) assay was conducted. The antifouling performance of the membranes was evaluated using a static protein adsorption assay, as shown in Figure 9. The pristine cellulose acetate (CA) membrane exhibited a high relative protein adsorption (RP%) of approximately 79%, indicating a pronounced tendency for fouling. Following grafting with CS/PVA/CA-RO fibers, the RP% significantly decreased to around 15%, reflecting a marked enhancement in antifouling properties. In comparison, typical membranes achieve only moderate reductions in protein adsorption and continue to show high platelet adhesion. The substantial reduction in protein adsorption can be attributed to two main factors. First, surface chemical modification: the incorporation of amino (–NH2) and hydroxyl (–OH) groups within the CS/PVA layer increases membrane hydrophilicity, promoting the formation of a stable hydration layer. This hydrated layer functions as both an energetic and physical barrier against protein adsorption, hindering direct contact and adsorption of protein molecules onto the membrane surface.
44
Second, surface morphology adjustment: the incorporation of CS/PVA fibers modifies charge distribution across the surface, creating electrostatic repulsion between the membrane and negatively charged protein species
45
The combined effect of these chemical and structural improvements results in a highly stable, low-fouling surface. Such properties make the CS/PVA/CA-RO composite membrane particularly advantageous for long-term operation in water purification and biomedical filtration systems, where resistance to organic fouling is essential for maintaining efficiency and durability. Illustrates the static protein adsorption measured on the pristine CA membrane and the CS/PVA/CA-RO composite membrane.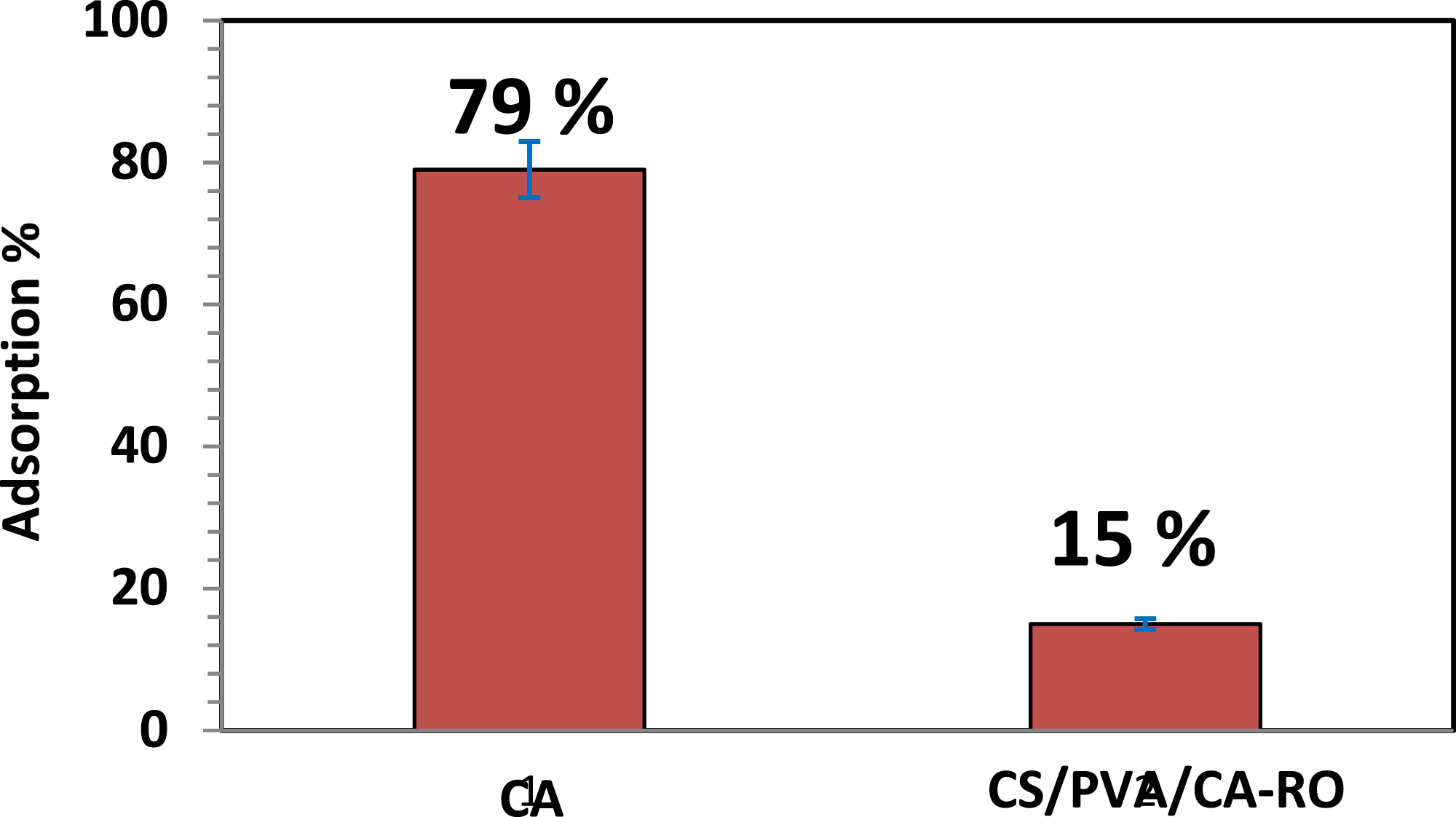
Conclusions
This study successfully demonstrates the fabrication of electrospun chitosan/poly (vinyl alcohol) (CS/PVA) fibers and their grafting onto cellulose acetate (CA-RO) membranes to produce biopolymer composite membranes with enhanced mechanical and antifouling properties. The electrospun CS/PVA fibers exhibited diameters ranging from 182 to 459 nm, where higher electrospinning voltages resulted in smaller and more uniform fibers, reflecting improved macromolecular chain stretching and fiber formation. Structural and morphological analyses confirmed that grafting CS/PVA fibers onto the CA-RO membrane enhanced surface roughness and porosity while maintaining a predominantly amorphous structure, which is favorable for membrane flexibility and permeability. Surface modification significantly improved membrane hydrophilicity, as evidenced by a decrease in water contact angle from 68.21° for pristine CA to 51.2° for CS/PVA/CA-RO membranes. In parallel, mechanical performance was markedly enhanced, with tensile strength increasing from 86.17 MPa to 144.55 MPa, attributed to strong intermolecular hydrogen bonding and improved macromolecular compatibility between chitosan, PVA, and cellulose acetate. The CS/PVA/CA-RO composite membranes exhibited excellent functional performance, achieving high rejection efficiencies of 85.98% for Pb(II), 90.51% for Cu(II), and 95.31% for Cd(II), with only a marginal change in water flux. These results indicate that the synergistic combination of electrospun CS/PVA fibers and surface grafting effectively integrates adsorption functionality with mechanical reinforcement and antifouling behavior. This work highlights a robust macromolecular engineering strategy for designing functional biopolymer composite membranes by correlating polymer structure, intermolecular interactions, and macroscopic performance. The findings provide valuable insights into structure–property relationships in chitosan-based systems and demonstrate their potential for advanced separation and antifouling membrane applications.
Footnotes
Authors contributions
Thria Alkhaldi, Ahmed H. Abdel-Salam and Zarah Alqarni: Conceptualization, Writing & Data collection, Asmaa Mohamed and Ashraf Morsy: Writing, editing & Software.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the University of Jeddah, Jeddah, Saudi Arabia, under grant No. (UJ-25-DR-1134). Therefore, the authors thank the University of Jeddah for its technical and financial support.
Data Availability Statement
Data will be made available on request.