
Abstract
Polyurethane/graphene composites have attracted significant attention featuring their wide applications in automotive interiors, coatings, and leather parts to bioengineering. This research focuses on the facile synthesis of biobased polyurethane (PU) by employing sunflower oil as a renewable resource. The synthesized biobased PU was reinforced with graphene nanoplatelets (GNP) and graphene nanoribbons (GNR) to fabricate composite films by solution-casting technique. By changing the filler content (0.01-0.05 wt%), a series of composite films were prepared. The addition of graphene derivatives to the PU matrix improved the mechanical, thermal, and surface properties of the composites compared to the pristine PU film. The incorporation of GNR and GNP fillers enhanced the storage modulus of polymeric film, as for neat PU, was noted maximum at 892 MPa which increased and noticed a maximum of 1121 MPa and 1487 MPa for 0.02 wt % GNP/PU and GNR/PU composite films, respectively. The tensile strength was obtained at 35 MPa and 14 MPa for 0.02 wt % GNP/PU and 0.01 wt % GNR/PU, as compared to 9.45 MPa for the pure PU film. The morphological studies by atomic force microscopy showed a good distribution of GNR and GNP fillers within the polyurethane matrix. The GNP/PU and GNR/PU composite films displayed excellent chemical resistance to different solvents. This work put forward a great utilization of renewable sunflower oil and graphene derivatives to fabricate PU composites with improved properties for potential applications.
Keywords
Introduction
Polymer composites are widespread materials with a range of increasing applications day-to-day. These materials play a major role in the industries such as packaging, aircraft, automobile, and construction sectors.1,2 In recent days, polyurethane (PU) has attracted researchers as a polymer for composites preparation because of its exceptional properties such as durability, low-temperature flexibility, and resilience as well as excellent adhesion property.3,4 PU composites can be utilized to prepare functional polymer coatings, solar cells, adhesives, synthetic leather, and sealants.5,6 PU is formed by the reaction between isocyanate (-NCO) and a polyol (a compound containing hydroxyl groups, -OH), where the isocyanate group and polyol represent the hard segment and soft segments, respectively. 7 However, the raw materials used in the preparation of PU are mainly from petrochemical resources and are toxic to the environment. This utilization of resources leads to the depletion of petroleum products. So, research is switching towards the use of biobased polyols using natural oils for PU synthesis.8,9 In addition, there are several chemical approaches to convert renewable oils into biobased polyols, such as hydroformylation, 10 epoxidation/ring-opening,9,11 thiol-ene coupling12,13 etc., to make them suitable for PU synthesis.
Vegetable oil-based polymer composites with graphene and its derivatives.

There are several reports on the incorporation of graphene and graphene oxide (GO) into polymer composites for improved barrier or electroactive application.17,25,26 But the uniform dispersion into a polymer and its high manufacturing costs stills remain a challenge for widespread application. 27 Thus, graphene nanoplatelets or graphene nanoribbons (GNR) have recently emerged as new ideal fillers for polymer reinforcement.28–31 GNP is a 2D structure consisting of multilayered graphite planes. 31 On the other hand, GNR are strips of graphene usually synthesized by the unzipping of CNT. 32 It has various structures based on length and width, such as a chiral, zigzag, armchair, etc. 33 Because of their high carbon-aspect ratio, GNR are good for application in conductive polymer composites and energy storage devices. For example, Pinto et al. 34 reported reduced gas permeability by 65% and 60% in the GNP and GO-reinforced poly (lactic acid) composites. The tensile properties of the GNP/epoxy composites gradually increased with the inclusion of GNP (up to 6 wt %), as revealed by King et al. 28 Low-defect GNRs and thermoplastic polyurethane (TPU) based composite films by solution casting for improved gas barrier and mechanical performances was also reported. 35 Although, there are several claims on the development of GNP or GNR-based polymer composites, the research on the fabrication of GNP or GNR-based PU composites for high-performance applications is still lagging.
In the present study, our approach is to prepare polyurethane from bio-based sunflower oil via epoxidation followed by a ring-opening mechanism. During the process, graphene derivatives (GNP or GNR) were dispersed in the polyol by the ultrasonication method to fabricate novel polyurethane composites with improved stiffness and strength. GNR used in this study are synthesized by unzipping multi-walled carbon nanotubes (MWCNT). The effects of GNP and GNR contents on the film formation, mechanical strength, thermal stability, and morphological properties of the composites were investigated. The incorporation of GNP or GNR (with increasing content) improved the hydrophobic property of the PU/GNP and PU/GNR composites, as determined by the water contact angle measurements. In addition, PU composites displayed excellent chemical resistance properties toward certain solvents. However, the mechanical properties of the composites were partially improved, attributed to the interfacial interaction and uniform dispersion within the GNP or GNR and polymer matrix. Therefore, this work provides a versatile approach for the preparation of bio-based PU composite films for potential applications in different sectors.
Materials and methods
Sunflower oil was obtained from a local Walmart (Pittsburg, KS, USA). Methylene diphenyl diisocyanate (MDI, RUBINATE®) was gifted from Huntsman (The Woodlands, TX, USA). AmberLite IR120H resin was procured from Alfa Aesar, USA. Lewitt MP64 ion-exchange resin, acetic acid (99.7%), sodium sulfate, ethyl acetate (99.9%), hydrogen peroxide, toluene, phosphoric acid, and methanol (99.9%) were purchased from Fisher Scientific, USA. GNP and MWCNT were purchased from Sigma Aldrich, USA. GNR were synthesized from MWCNT. Hydrochloric acid, sulfuric acid, and potassium permanganate were purchased from Acros Organics, USA. For testing, Pro Force SAE 5W-30 motor oil purchased from a local Walmart, Pittsburg, Kansas was used. Diesel was obtained from a local gas station, Pittsburg, Kansas.
Synthesis of polyol from sunflower oil
A two-stage reaction process following epoxidation and the ring-opening procedure was adopted for the synthesis of polyol (Figure 1). In the epoxidation of SO, the molar ratio of double bond/acetic acid/hydrogen peroxide was kept at 1: 0.5: 1.5. For the process, 300g of sunflower oil, 75g of AmberLite IR120H resin as a catalyst, and 150 mL of toluene (50 wt% of oil) were poured into a three-necked round-bottom flask. The mixture was stirred for 15 min at 5°C–10°C. Then, 43.9 mL of acetic acid was added by a dropping funnel followed by stirring for 30 min. At the same reaction condition, 180 mL of 30% H2O2 was further added dropwise to the solution flask. After the complete addition of H2O2, the temperature was increased to 70°C and stirred for 7 h. Then, the reaction mixture was cooled down to room temperature and the resin was drained out by filtration. The pH of the mixture was determined. After that, the solution was washed thoroughly with 10% brine solution. Finally, the solution was concentrated by a rotary evaporator operating at 70°C. The final product obtained was epoxidized sunflower oil (ESO) with a conversion of about 89%. The synthesized ESO was further used for the preparation of polyol following the epoxy ring-opening mechanism. Preparation of sunflower oil-based polyol following epoxidation and ring-opening mechanism.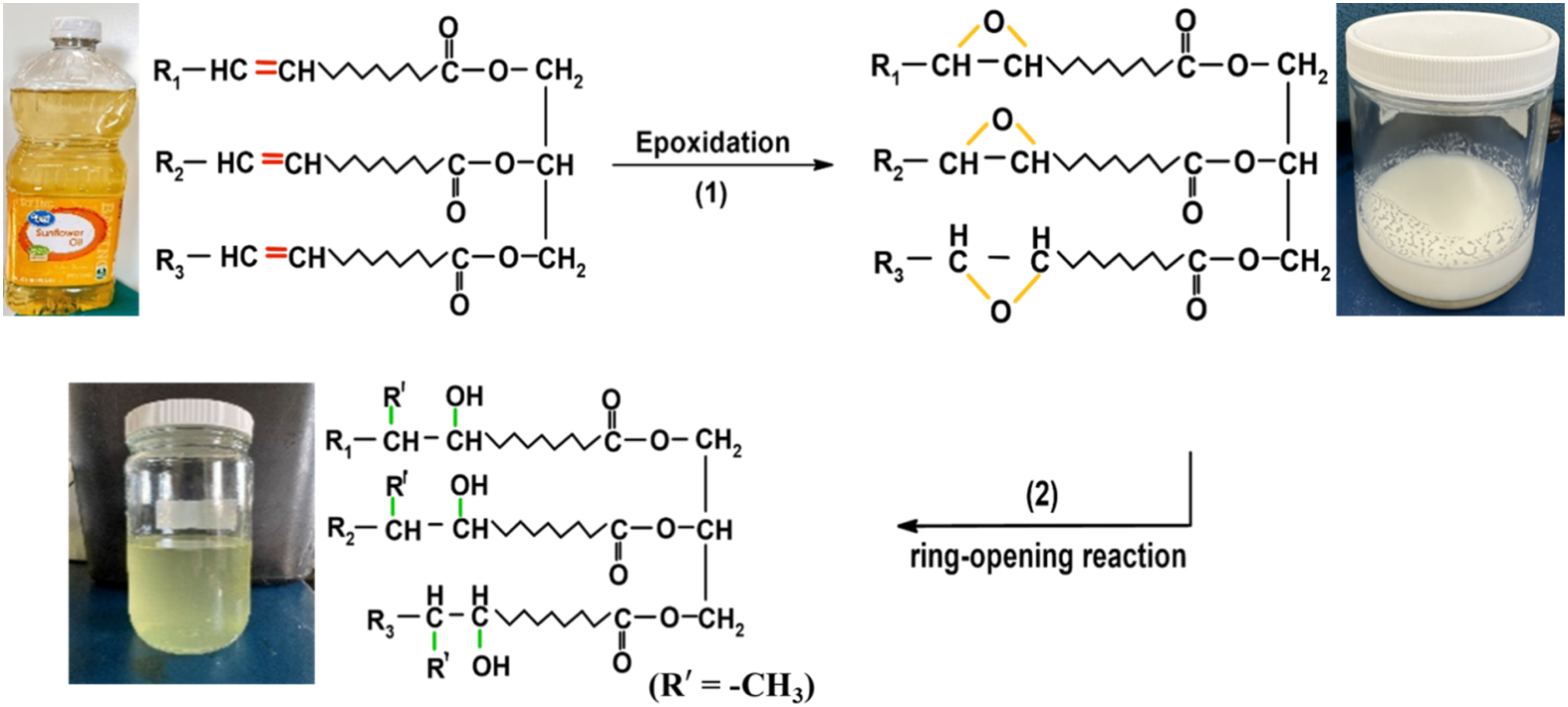
For the ring-opening reaction, a three-necked flask equipped with a dropping funnel and condenser was taken. In the reaction flask, methanol (7: 1 M ratio to the epoxy group), and tetrafluoroboric acid (0.05 wt% of ESO and methanol) were mixed and the temperature was set at 70°C. Then, ESO was added to the mixture via a dropping funnel and stirred continuously for 70 min. The mixture was further refluxed for 60 min, after the complete addition of ESO. This step was performed in the presence of a condenser to prevent the evaporation of methanol. Then, the reaction solution was cooled down and Lewatit MP64 ion-exchange resin was added to the flask for neutralization with stirring for 30-45 min. The pH of the solution was checked after the resin was filtered. A both high and low vacuum was applied for 2 h for complete evaporation of methanol. The resulting yield of sunflower oil-based polyol was obtained at around 82%.
Preparation of graphene nanoribbons
MWCNT were utilized for the fabrication of GNR. In the synthesis procedure, 1g of MWCNT was dispersed in 32 mL of phosphoric acid (H3PO4) and 280 mL of sulfuric acid (H2SO4) by using a magnetic stirrer. After mixing for a few minutes, 10g of potassium permanganate (KMnO4) was added and stirred at 65°C for 4 h. The resulting mixture was then cooled to room temperature and poured over a mixture containing 800 mL ice-water and 40 mL hydrogen peroxide (30% H2O2). Further, the mixture was left overnight and then filtered using 0.2 µm mesh PTFE filters. The solution was washed with water and ethanol. The black-colored compound obtained was dried in a vacuum oven at 65°C for 10 h and utilized as filler for PU composite preparation. The typical synthesis route for GNR is presented in Figure 2. Schematic diagram of GNR synthesis route.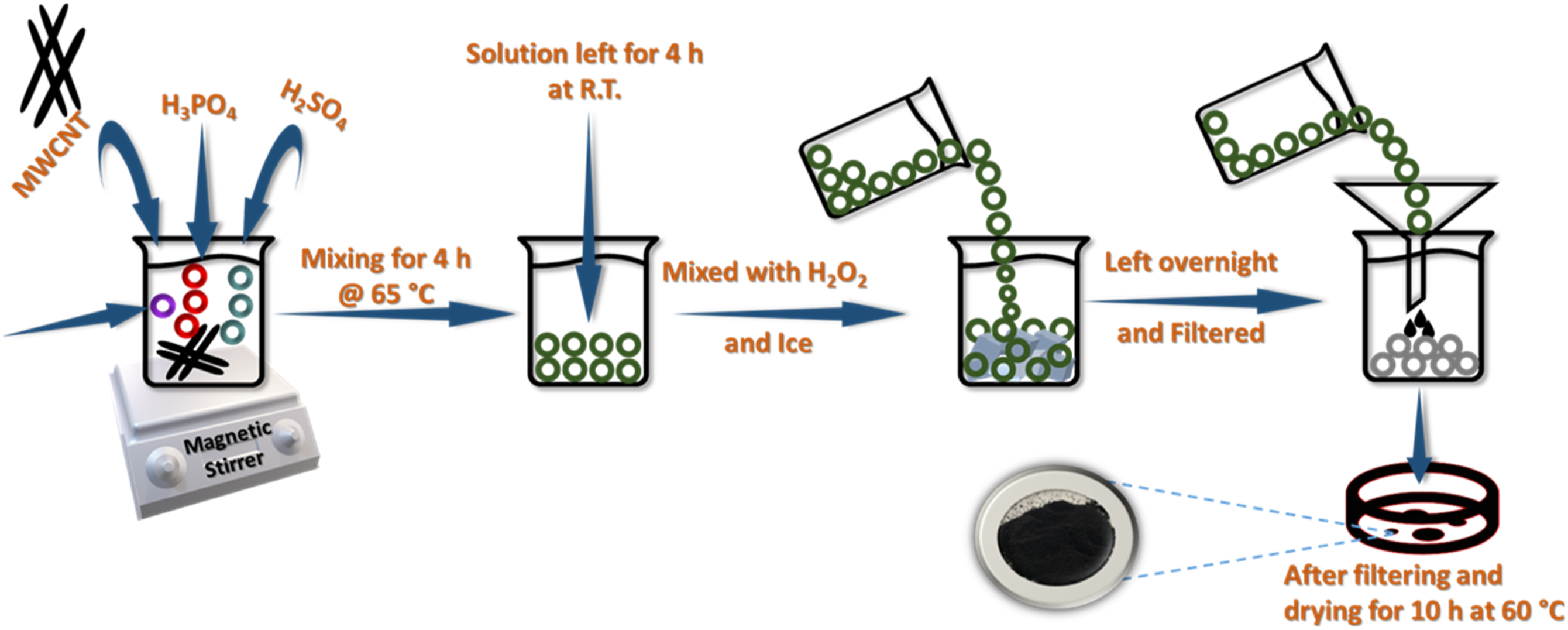
Preparation of PU/GNP and PU/GNR composite films
The synthesized sunflower oil-based polyol was mixed with GNP or GNR separately and effectively dispersed using an ultrasonic bath sonicator (Figure 3). For the preparation of composite films, 12.5g of polyol and 6.0g of isocyanate along with different amounts of fillers (0.01, 0.02, and 0.05 wt% to polyol) were used. GNP or GNR were dispersed into polyol solution for 2 h. After that, methylene diphenyl isocyanate (1:1 M ratio to polyol) was added to the graphene-dispersed polyol. After mixing for 10 min, the solution was poured into a Teflon petri-dish for film formation. Finally, the sample was cured in a vacuum oven for 90 min at 70°C. Then, rectangular strips of specific dimensions were cut from the cured films and used for different analyses by standard testing methods. The pictures of the PU/GNP and PU/GNR cast films with variable composition are shown in Figure 4. A schematic illustration for the fabrication of PU/graphene-derivative composites. Photographs of different PU/GNP and PU/GNR composites.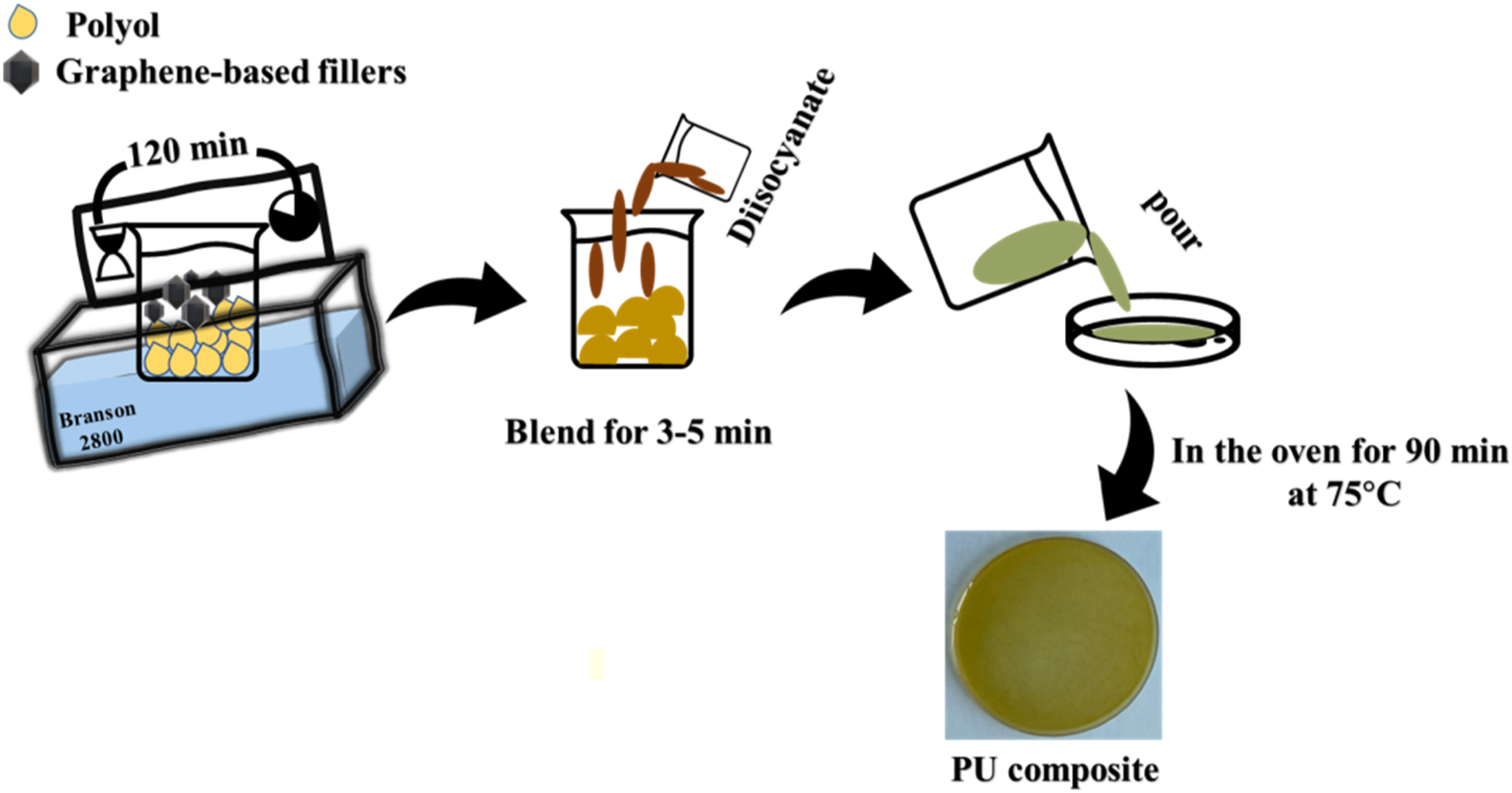

Characterization
Physical properties
FT-IR spectra were recorded using a PerkinElmer Spectrometer in transmission mode (4000-500 cm−1). A total number of 32 scans with 4.0 cm−1 resolution were followed. Waters gel permeation chromatography (GPC) instrument equipped with 515 HPLC pump was used to measure the molecular weight of samples. Tetrahydrofuran (THF) was used as the eluent at a flow rate of 1 mL/min and polystyrene was used as a calibration standard. The oil and polyol viscosity were examined by TA instruments AR 2000 dynamic stress rheometer. Hanus method (IUPAC 2.205) was used to calculate the iodine value. The hydroxyl value (-OH value) of polyol was determined by the P.A.P method (IUPAC 2.241). ACS PER-OXI standard method was used to determine the epoxy content (% EOC) of ESO and polyol. GNRs and GNPs samples were characterized by a Shimadzu X-ray diffractometer (XRD) using Cu-Kα radiation (λ = 1.419 nm). The samples were scanned from 2θ = 1° to 80° at a rate of 1°min−1. The water contact angle (WCA) of the droplet was measured by the Ossila contact angle goniometer.
Thermal properties
Differential scanning calorimetry (DSC) testing of PU composite samples was conducted by a DSC Q100 from TA instruments. The samples were heated thermally from -80°C to +250°C at a 10°C/min heating rate. The glass transition temperature (T g ) was recorded from the DSC curves taking the baseline tangents using TA Universal Analysis software. Thermogravimetric analysis (TGA) was performed using TGA 550 (TA instruments) under the nitrogen atmosphere in 30 to 600°C temperature range at 10°C/min heating rate. Dynamic mechanical analysis (DMA) of the film samples was performed using Q800 TA instrument. The samples were tested under a temperature ramp from -50 to +150°C at a rate of 3°C/min, 1 Hz frequency, and amplitude of 10 μm.
Mechanical properties
The tensile properties of the composite films were measured using an Instron 3367 instrument at room temperature and at a crosshead speed of 500 mm/min. The flexibility of the composite films was tested using an MTS Qtest, Sintech flexural instrument as per the ASTM D790 test method. An average value of three replicates specimens from tensile and flexural strength analysis were reported. The hardness of the composite was analyzed by the PTC instrument type D durometer following the ASTM D2240 test method.
Morphological properties
The dispersion of graphene-based derivatives in the PU matrix was examined via atomic force microscopy (AFM) instrument (AFM Workshop, USA).
Results and discussion
FT-IR and GPC analysis of ESO and SO-based polyol
The structural change in functional groups during the synthesis of polyol from SO via epoxidation was analyzed by FT-IR spectra (Figure 5(a)). The absorption peak at 3010 cm−1 corresponding to =CH stretching in the SO disappeared in the FT-IR spectrum of ESO. Also, a signature peak of the epoxy ring vibration at 828 cm−1 appeared in the ESO which was absent in SO, which confirms the successful epoxidation of SO. In the case of the SO-based polyol spectrum, the epoxy ring vibration peak vanishes suggesting the utilization of the epoxy group for the ring-opening process. Moreover, a broad absorption peak at 3481 cm−1 was noticed in the polyol due to the formation of the hydroxyl (-OH) group. This confirms that the SO-based polyol was successfully synthesized from ESO. (a) FT-IR spectra and (b) GPC chromatograms of SO, ESO, and polyol.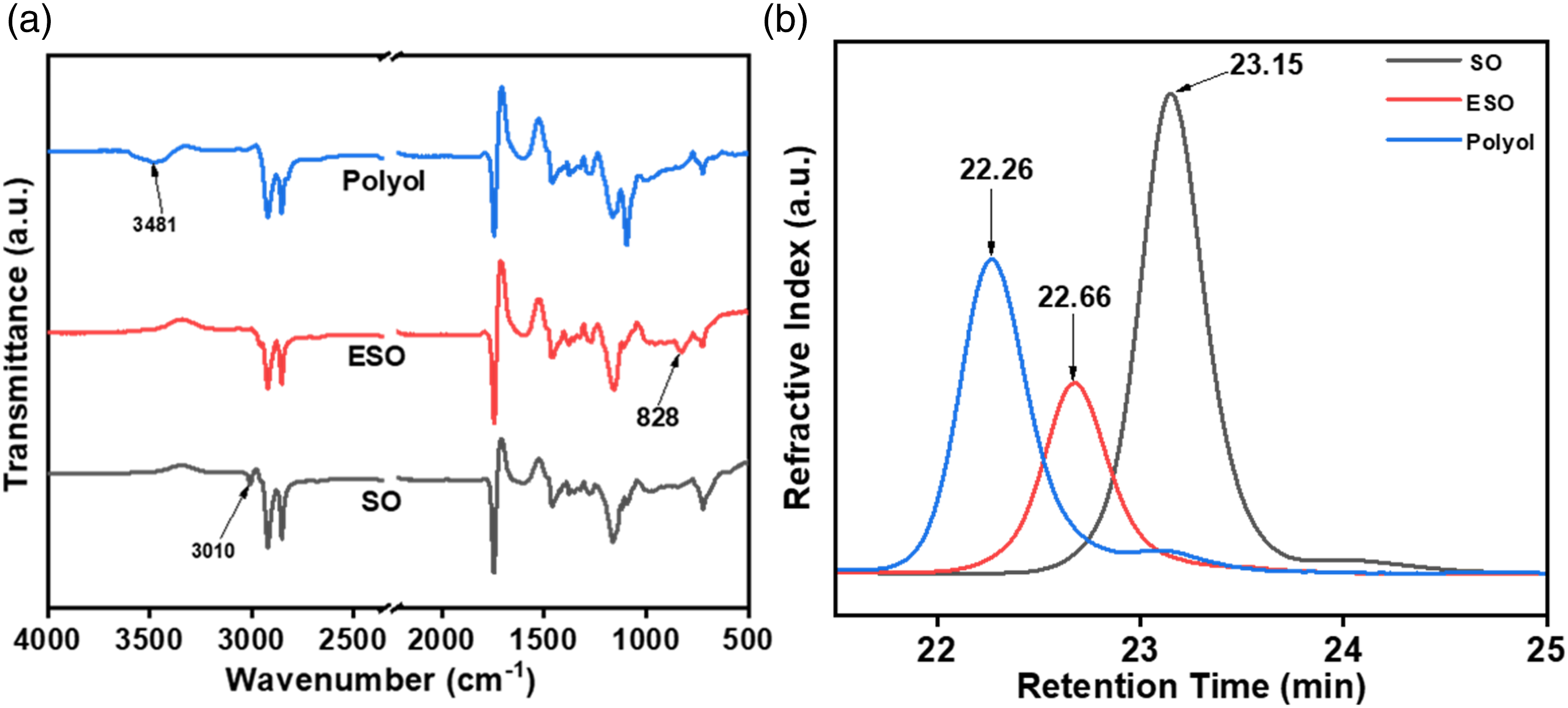
Figure 5(b) shows the GPC chromatograms for the successful conversion of SO into ESO, and SO-based polyol as a function of retention time. For the SO, the retention time was observed at around 23.15 min, whereas the retention time for ESO was obtained at around 22.68 min. The decrease in the retention time is directly related to the molecular weight of the compound. The lower the retention time, the higher is the molecular weight of the material.36,37 Furthermore, the synthesized SO-based polyol showed a retention time of 22.27 min, which indicates an increase in the molecular weight and relates to the formation of polyol from SO. The iodine value of the sunflower oil was obtained as 105g I2/100g, following the Hanus method. The epoxy content of ESO was obtained at 5.23%. The hydroxyl value (-OH value) of polyol was evaluated as 189 mg KOH/g.
XRD analysis of GNP and GNR
XRD spectroscopy was used to characterize the graphene derivatives. Figure 6 shows the XRD pattern of GNP and GNR. A sharp diffraction line at 2θ = 26.6° is observed for GNP. The nanoplatelets exhibited a highly crystalline graphite-like structure, with the presence of strong (002) and (004) reflections. On the other hand, GNR showed a relatively broad and low-intensity peak. This confirms the exfoliation and successful synthesis via unzipping of MWCNT.38,39 XRD patterns of GNP and GNR.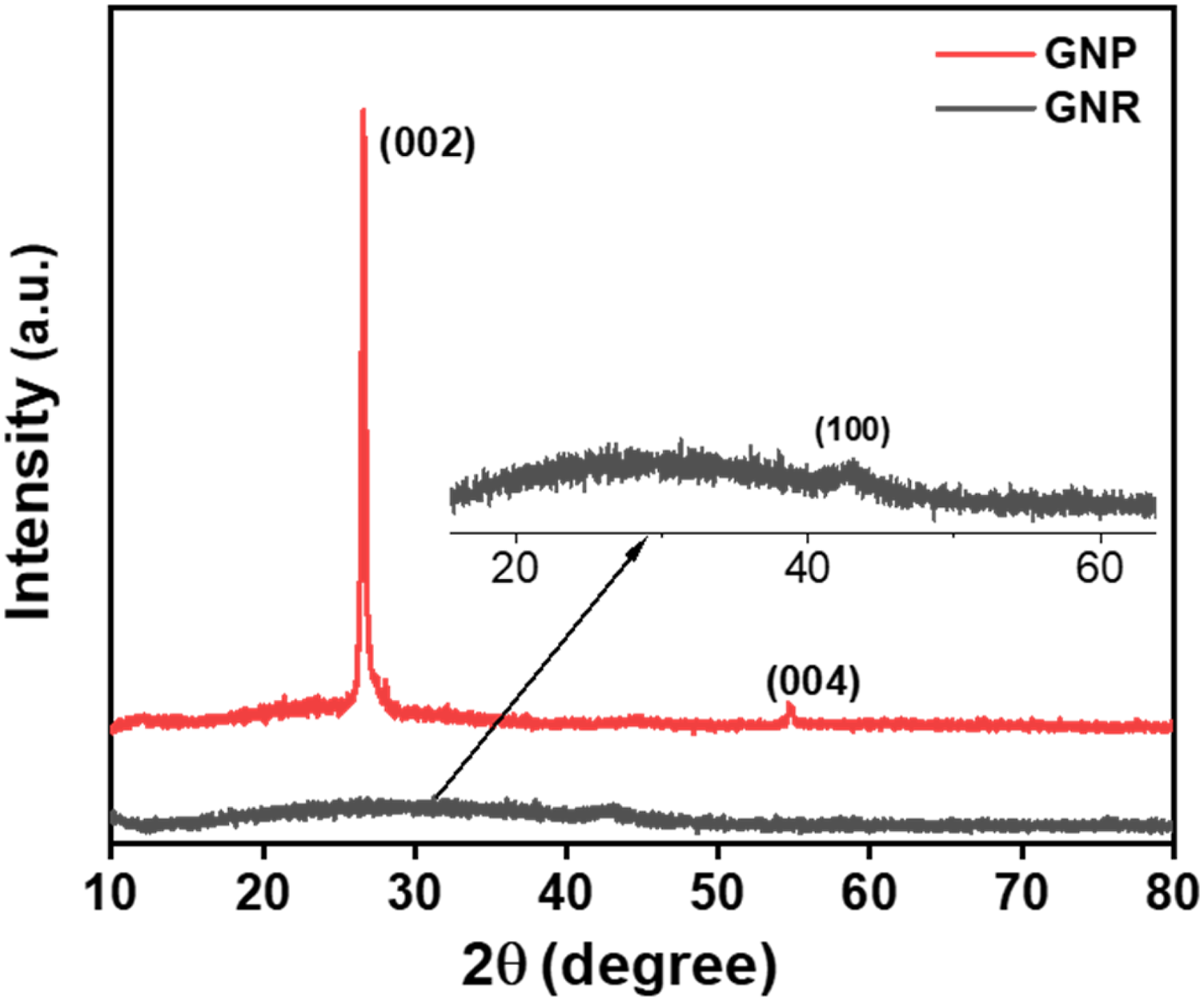
Dynamic mechanical analysis
The analysis was carried out from -50 to 150°C and the temperature dependence of storage modulus and tan delta are shown in Figure 7. In the case of PU/GNR composite (Figure 7(a)), the storage modulus was higher for 0.02 wt% and 0.05 wt% GNR than pure PU film. The maximum storage modulus for composites with 0.02 wt% and 0.05 wt% GNR is 1488 MPa and 1155 MPa, which is higher than pure PU of 958 MPa only. This property improvement could be attributed to the synergistic effect of phase separation and the incorporated GNR. Moreover, the tan δ peak and the T
g
was lowered with increasing GNR loading in the composite (Figure 7(b)). Pure PU registered T
g
of 52.6°C, whereas 0.01 wt% GNR filled composite displayed T
g
at 50.4°C, and 0.02, 0.05 wt% GNR loaded composite showed similar T
g
around 40.0°C, respectively. Generally, the addition of fillers will increase the T
g
of the polymer to a higher temperature by restricting the polymer chain motion. However, the addition of GNR in the composite shifted the T
g
of the polymer to a lower temperature. This result could be due to the phase segregation of PU that included a few hard segments combined with soft segments. This leads to an effortless movement of the soft segments and the influence of the hard segment becomes more than the filler in T
g
determination. The phenomenon was also supported by the analysis of rubbery region of interest from the storage modulus plot (Figure 7(a)). With variation of GNR content, the storage modulus in the rubbery region of the GNR/PU composites were observed lower compared to pristine PU. A similar observation was reported in the earlier studies on TPU/carbon nanotube composites.40,41 Temperature dependence of (a) storage modulus, and (b) Tan delta of pure PU and GNR/PU composites.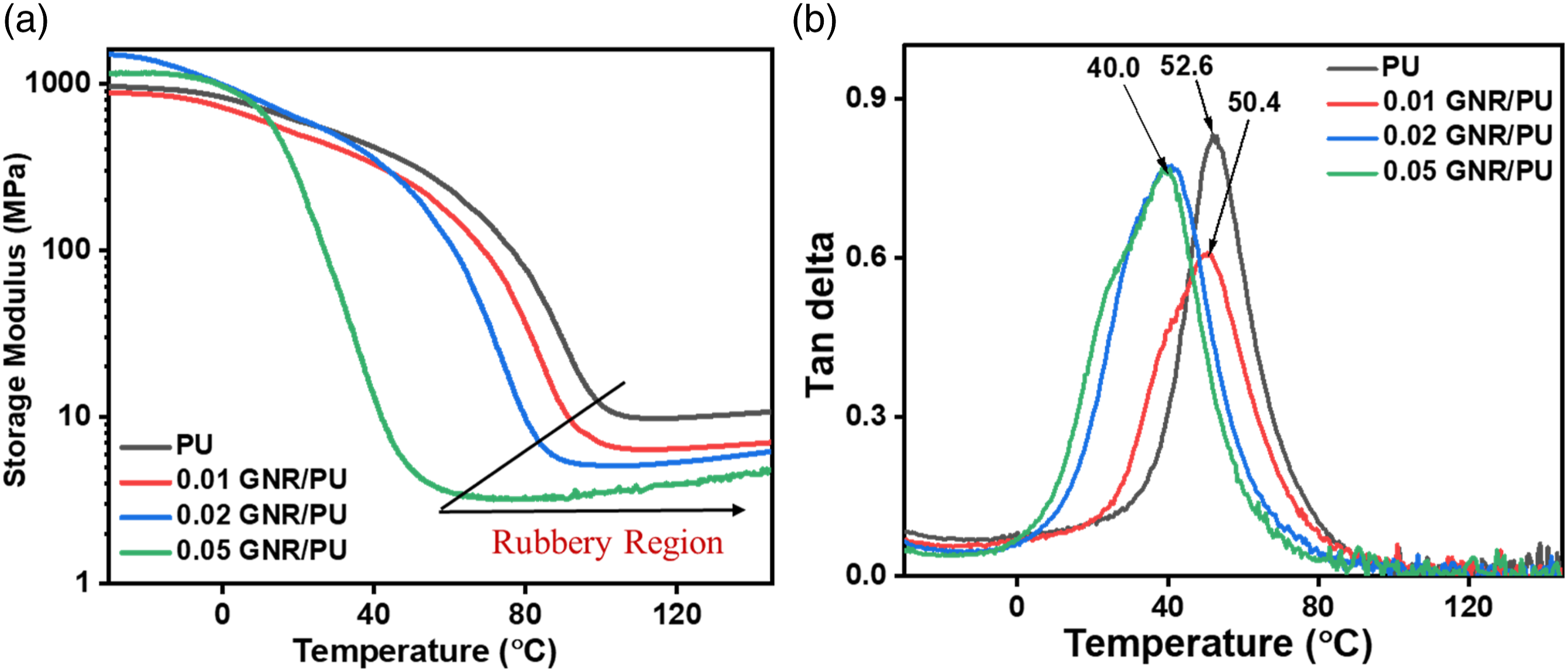
The storage modulus and tan delta plots versus temperature with different compositions of GNP/PU composites are shown in Figure 8. While adding 0.02 wt% of GNP, the storage modulus improved to that of pure PU. The maximum storage modulus for neat PU film was 958 MPa whereas for the 0.02 wt% GNP/PU is more than 1154 MPa. This advancement could be due to the reinforcement effect of GNP in the PU matrix. With the addition of higher GNP, the storage modulus for the composites decreased drastically as compared to the pure polyurethane. This could be due to agglomeration of the GNP particles, which at high concentrations loses it “nano” reinforcement tendency with the polymer phase. For the tan δ plots (Figure 8(b)), the incorporation of GNP in the composite lowered the damping factors, and the T
g
was increased with increasing content of GNP. The T
g
of the pure PU was obtained at 52.6°C, whereas 0.01, 0.02, and 0.05 wt% GNP-filled composite showed increased T
g
of the PU soft phase between 58.7 and 64.0°C, respectively. This could be attributed to the better reinforcement and interfacial interaction between the naturally occurring polar ethers, carboxyl’s or hydroxyls functional groups of GNP additive and PU matrix.
42
(a) Storage modulus versus temperature and (b) Tan delta versus temperature graphs for neat and GNP/PU composite.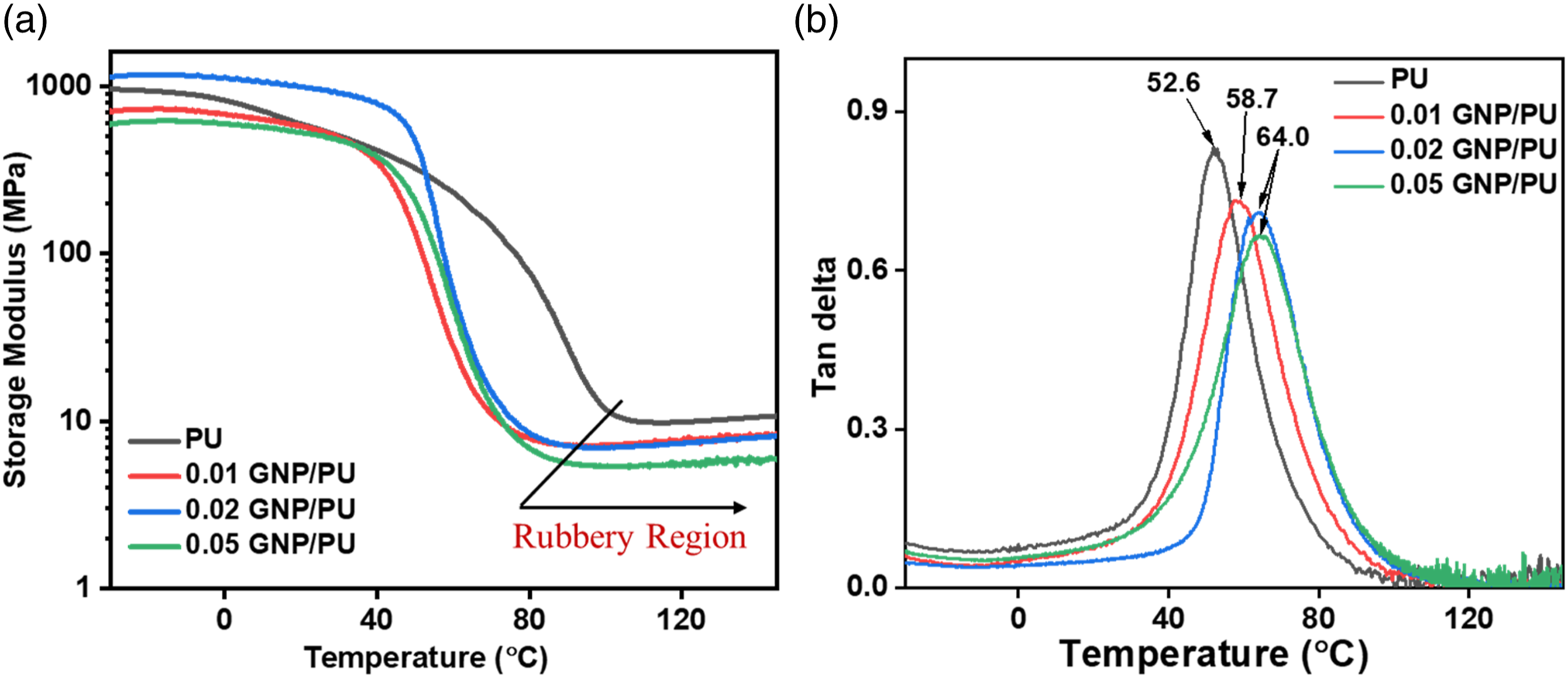
Thermal properties of PU composites
The thermal stabilities of the composite films by adding graphene additives (GNR and GNP) to the PU matrix were studied by thermogravimetric analysis (Figure 9). Pristine PU generally shows a two-step degradation pattern, mainly the hard segment (urethane linkages) decomposes first followed by the soft segments (polyols). The initial dissociation of urethane bonds results into primary amines, or olefins and carbon dioxide.43–45 In the case of GNR/PU composites, the change in thermal decomposition pattern was not significant and almost similar to pure PU. Considerably, all samples showed differences in the residual char yield percentage. 0.02 and 0.05 wt% GNR reinforced PU composite yielded around 9% char yield compared to 1.4% for the neat PU sample. This improvement could be because of the localized interaction of the GNR molecule with the PU matrix.
46
TGA curves of (a) GNP/PU composite and (b) GNR/PU composite samples.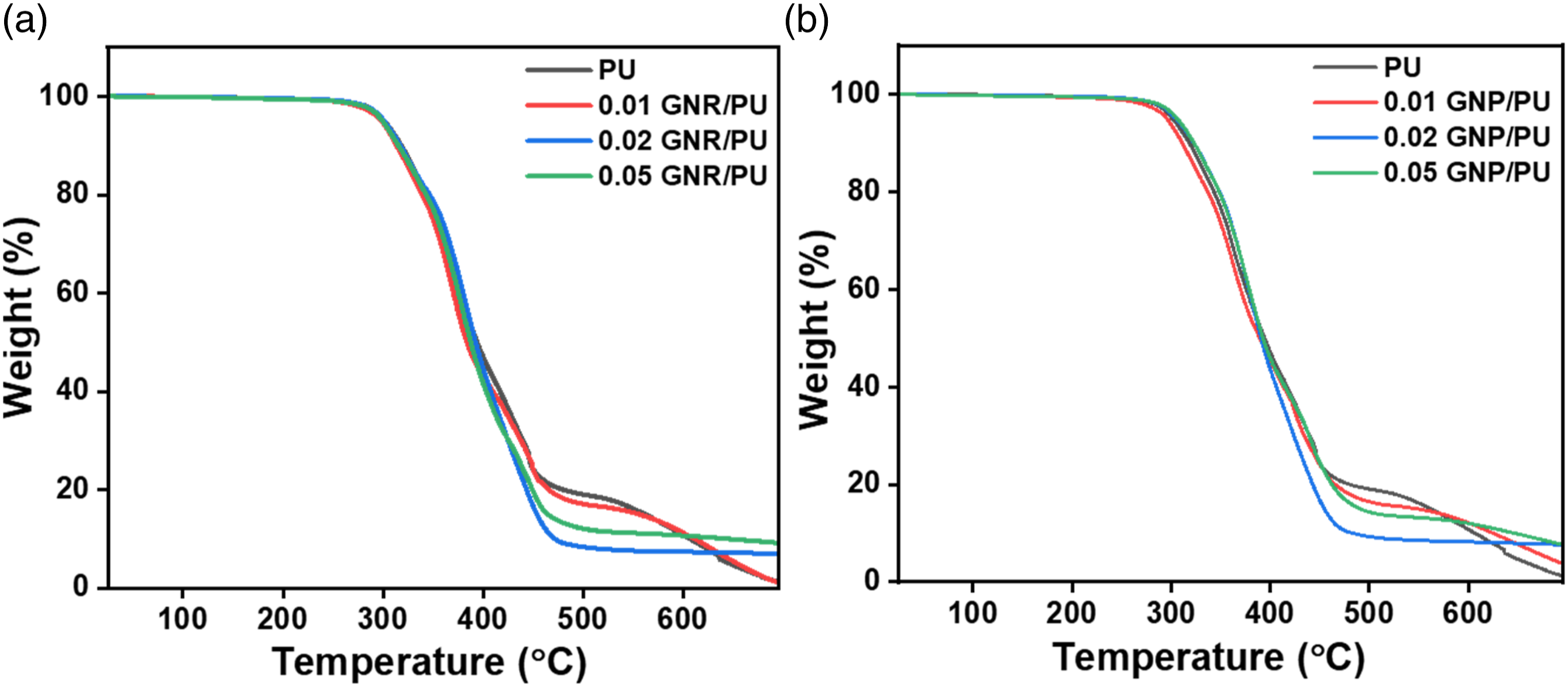
The enhancement of thermal stability was minimal with the addition of various concentrations of GNP, as shown in Figure 9(b). The first and second-stage degradation were observed around 280 and 460°C for neat PU and GNP/PU composite samples, respectively. It is worth mentioning that the residue formation was 1.4% of the total weight for neat PU, which increased after the addition of 0.01 wt% GNP and showed 3.9% residue. With the highest addition of GNP (0.05 wt%), the percentage of the left product was attained at 7.8% of the total weight. This indicates an almost 6.4% improvement in residue percentage for 0.05 wt% GNP/PU formed at a higher temperature than the neat PU. This improved thermal stability may be ascribed to the good interlinkage of the GNP surface with the PU matrix, which in terms enhanced the thermal properties of the PU composite. 47 The higher surface area of the graphene nanomaterial also provides a way for better thermal stability with higher degradation temperature and greater char yield. The maximum char yield residues are obtained in the range of 7 to 9%, similar for both GNR and GNP composites.
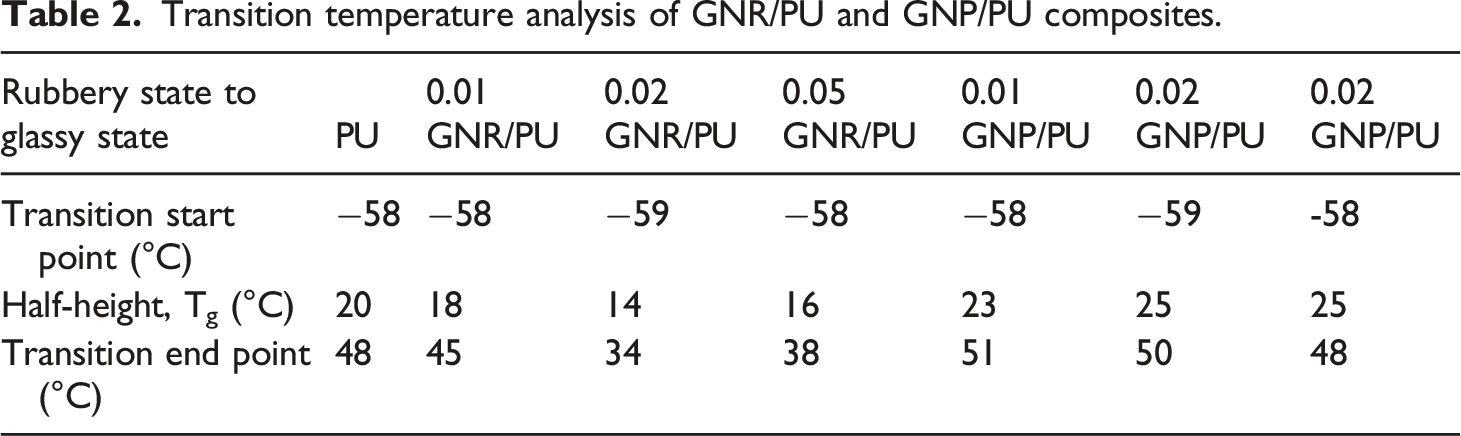
Figure 10(a) represents the DSC traces of neat PU and GNR/PU composites. The pristine PU displayed a glass transition temperature of 20°C and GNR composites displayed lower Tg than pristine PU. With the increasing content of GNR in the composite, there is no significant variation in the Tg of the material. Similar observations were observed in the DSC curves of neat PU and GNP/PU composites (Figure 10(b)). The GNP/PU samples demonstrated Tg around 23°C–25°C temperature, with varying GNP content. The transition temperature was observed very close to neat PU which may happen because of the agglomeration of graphene derivative in the PU matrix. A similar observation was also reported in various research papers, where the Tg was suppressed by the addition of SiC/CNT nanoparticles in the polymer matrix.41,48 The detailed transition temperatures and Tg observed from the heating curve are tabulated in Table 2. Due to the dynamic measurements associated with DMA testing, the Tg obtained from DMA are higher than that obtained from DSC analysis. DSC curves of (a) neat PU and GNR/PU composites, (b) neat PU and GNP/PU composites. Transition temperature analysis of GNR/PU and GNP/PU composites.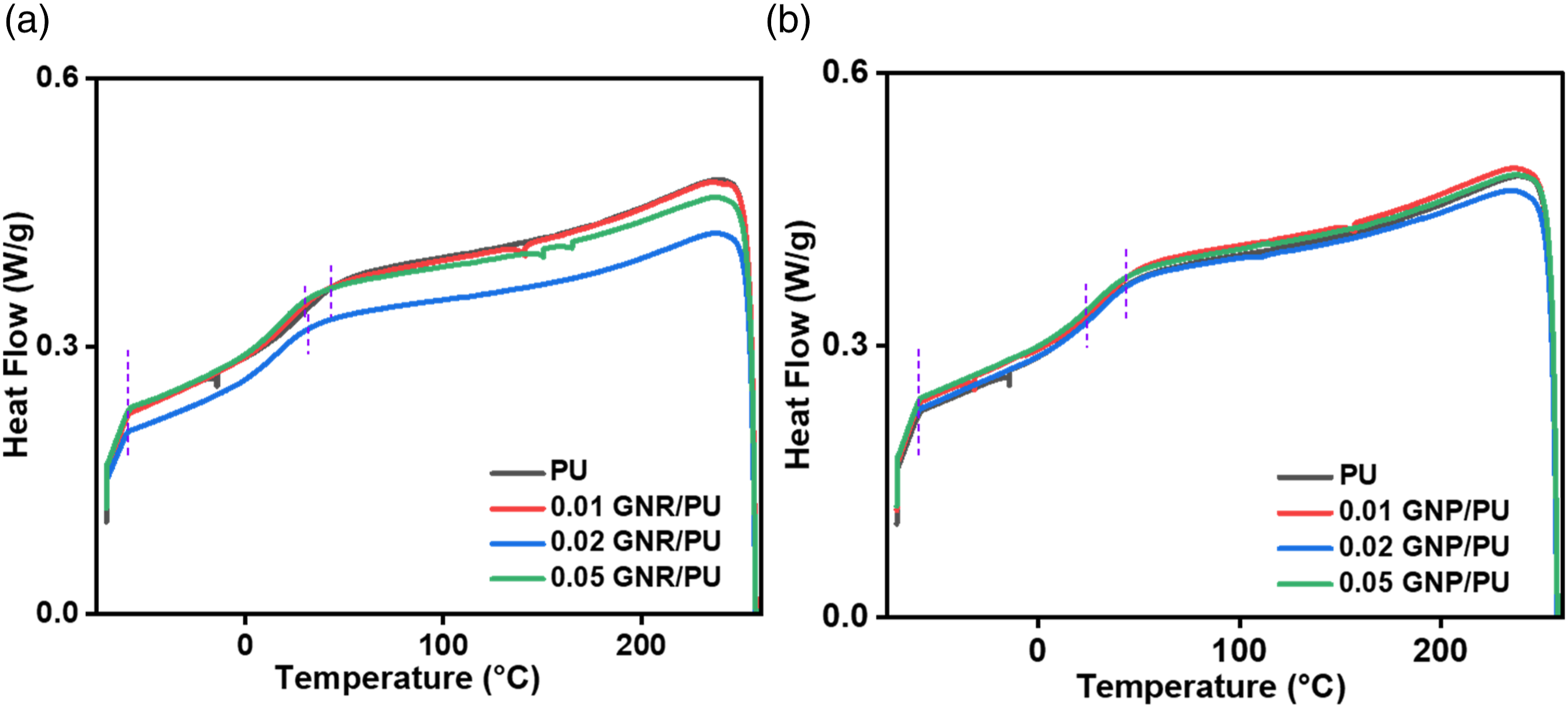
Mechanical properties
The tensile strength was enhanced with the addition of GNR and GNP in the polyurethane matrix, as shown in Figure 11. The tensile strength of the pristine PU corresponds to 9.47 MPa. The maximum tensile strength was observed at 13.9 MPa for the 0.01 GNR/PU composite, which is 40% higher than PU only. The yield point shifted toward higher values with the addition of GNR content i.e., for 0.02 wt% of GNR loading. The yield strength of 0.02 GNR/PU increased to 10.4 MPa, whereas decreases to 8.2 MPa for 0.05 wt% of GNR loading. The tensile strength of PU/GNR composites is not improved much due to poor interfacial adhesion between the polymer and the surface of GNR, resulting in the agglomeration of GNR particles. Tensile strength of neat and (a) GNR/PU composite films, and (b) GNP/PU composite films.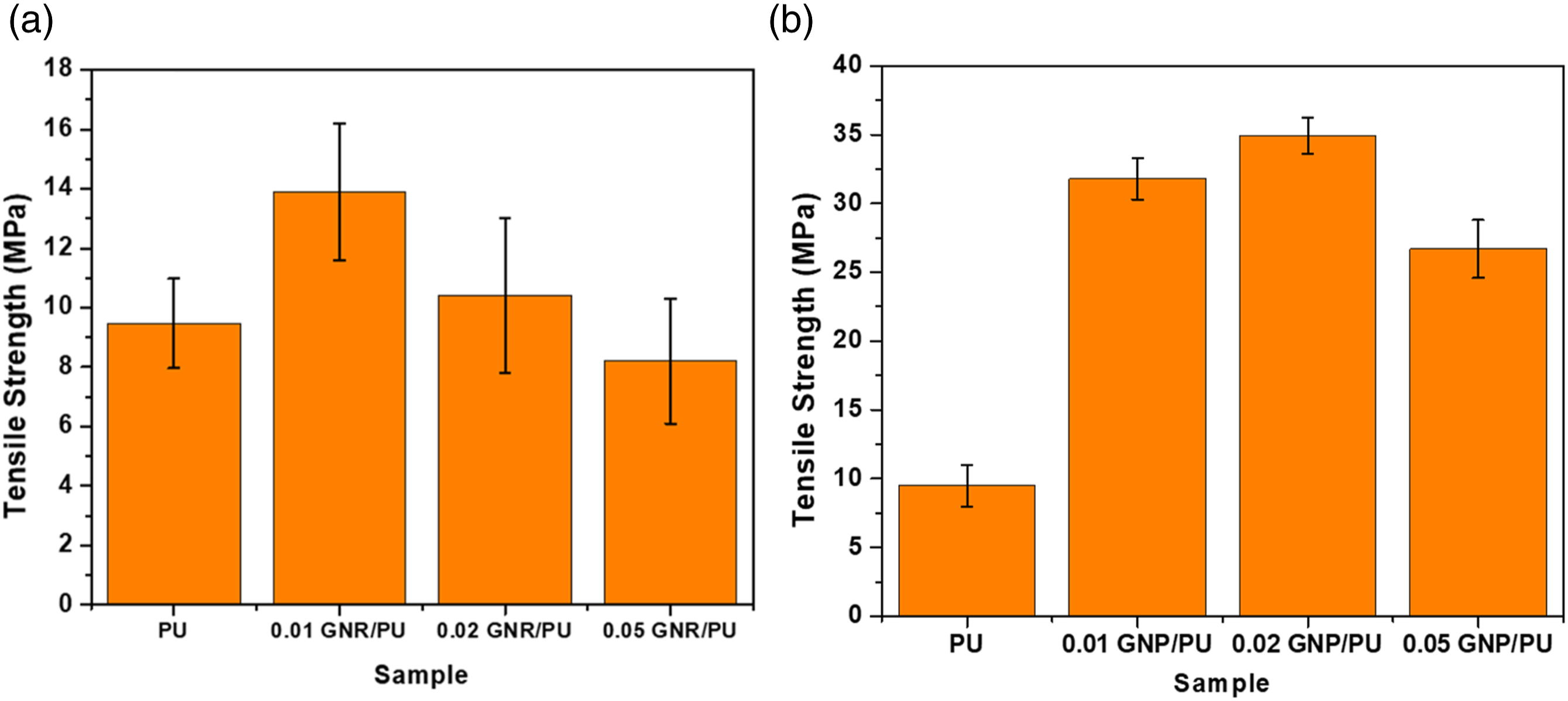
On the other hand, the PU/GNP composites showed a steeper slope than pristine PU suggesting the stiffness of PU/GNP composites is higher than neat material. 0.02 GNP/PU has the highest tensile strength of 34.9 MPa followed by 0.01 GNP/PU and 0.05 GNP/PU composites at 31.8 and 26.7 MPa, respectively. This means the introduction of GNP up to a certain concentration in the PU matrix resulted in the enhancement of the mechanical strength of composites. GNPs have more naturally occurring functional groups like ethers, carboxyl’s or hydroxyls than GNRs that can interact with the polyurethane matrix, showing better reinforcement, hence higher mechanical strength values. With higher loading, inferior dispersion and agglomeration of GNP layers resulted in the decrease of mechanical properties of the composites.
The flexural properties of the controlled PU and the composite films are presented in Figure 12. The controlled PU film showed flexural strength of 2.25 N and a vertical displacement of 5 mm. In the case of GNR/PU composite films (Figure 12(a)), the flexural strength increased to 12.0 N with a displacement of 12 mm for 0.01 wt% of GNR. With an increase in the GNR content to 0.02 and 0.05 wt%, the flexural strength of the films decreases but the displacement was observed highest at 18.0 mm. On the other hand, 0.01 and 0.02 wt% GNP-based films (Figure 12(b)) showed similar and maximum flexural strength and vertical displacement of 8 N and 8 mm, respectively. With further addition to 0.05 wt% GNP, the flexural strength gets lowered showing similar strength to the neat PU sample. Force versus extension curve of (a) GNR/PU and (b) GNP/PU composite films.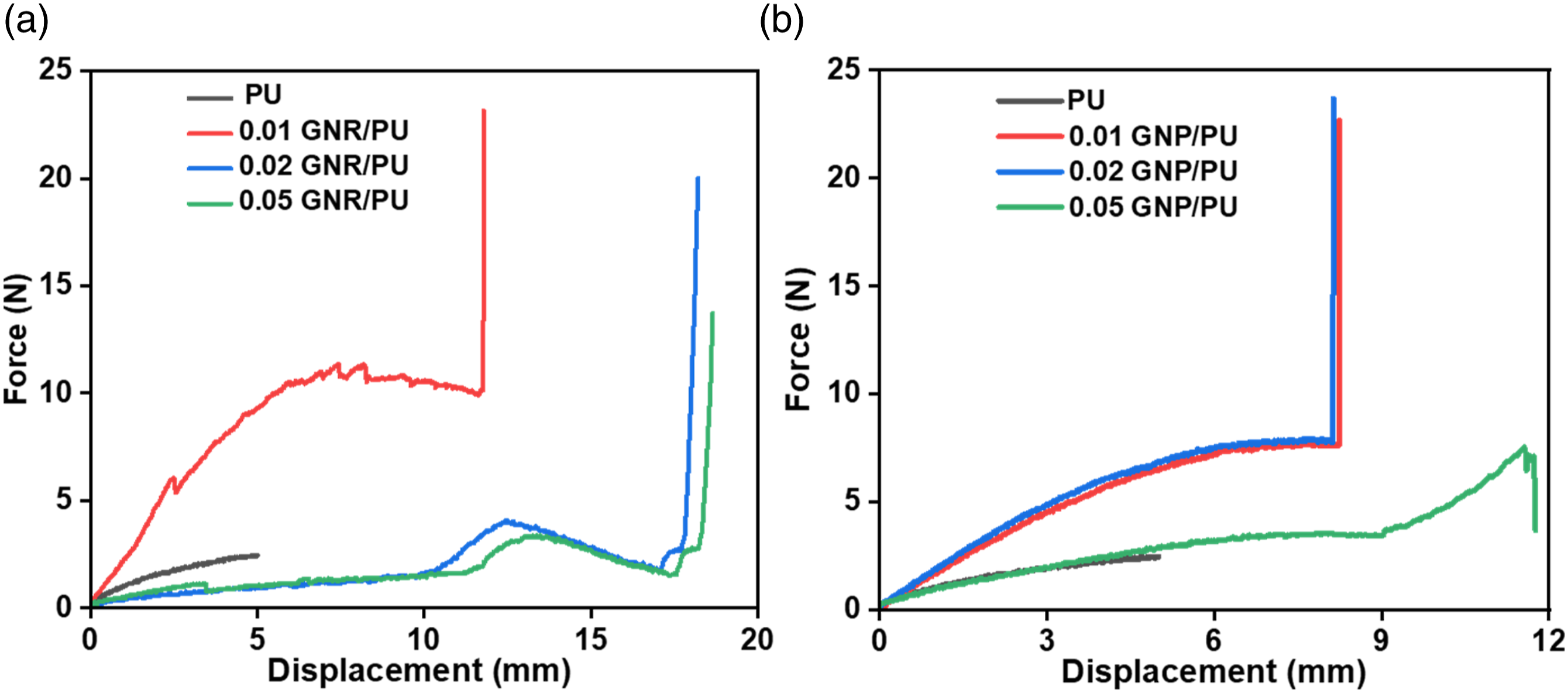
The Shore D hardness of the samples was investigated by ASTM D2240 standard test method. Figure 13 shows the average hardness values of the pure PU, GNR/PU, and GNP/PU composites. From the data, there was a continuous increase in the hardness of the composites with increasing content of GNP. The pure PU film, 0.01, 0.02, and 0.05 wt% GNP displayed hardness of 51, 62, 65, and 70 Shore D, respectively. The improved interfacial interaction between the urethane linkages and GNP sheets resulted in higher hardness values. Whereas the increasing trend of hardness is not continuous for the GNR-reinforced composites. 0.01 and 0.02 wt% GNR incorporated composites showed increased Shore D hardness of 62 and 58 compared to the pristine counterpart. The influence of the nanoparticle aggregation with reduction in mechanical properties is well-reported in some literature studies.17,49 Shore D hardness for pure PU and GNP, GNR/PU composites.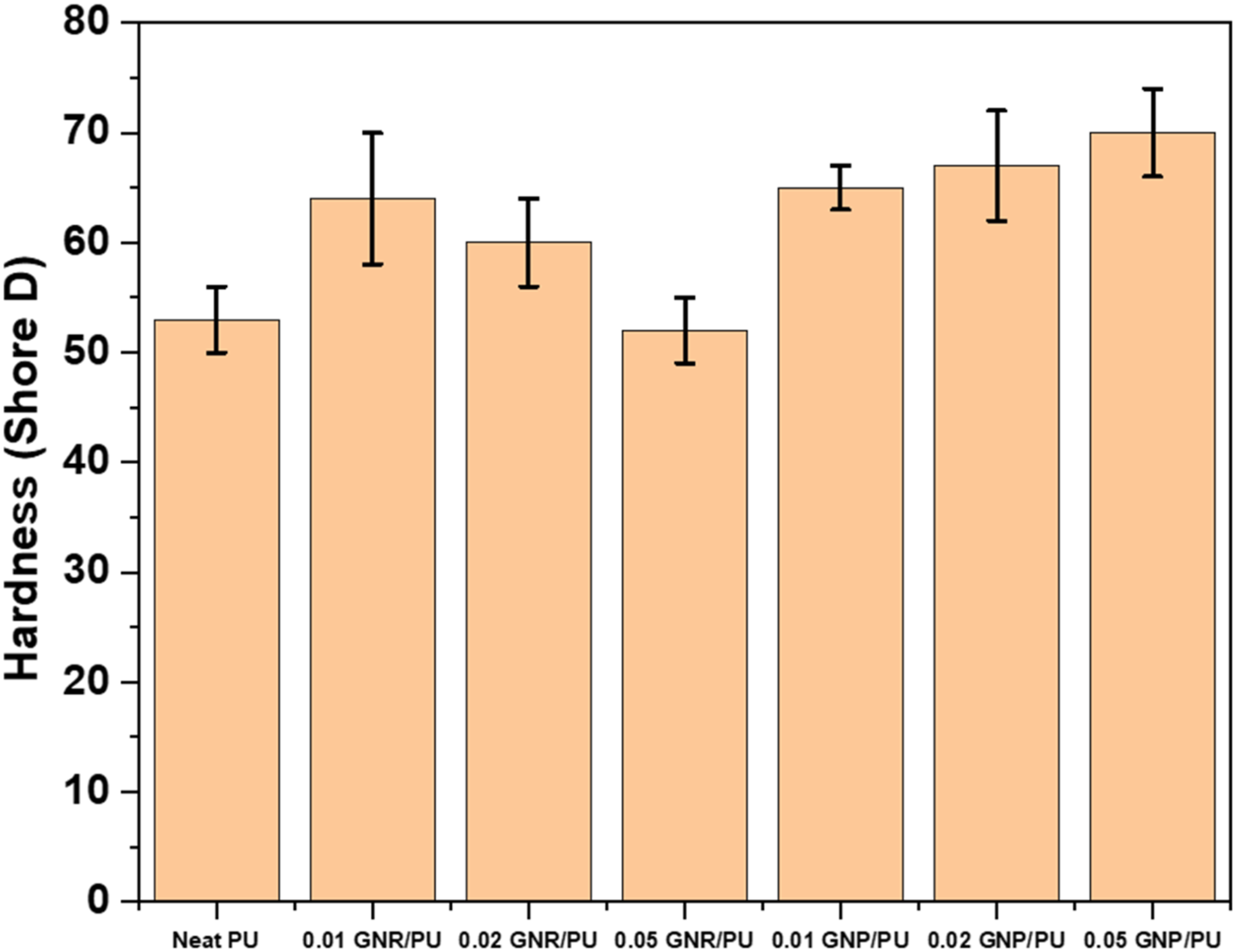
Water contact angle study
The wettability of the surface of a material is important for coating application. Therefore, the water repellency of the prepared PU film was studied by a contact angle goniometer using a droplet of 10 µL distilled water. The average contact angle data and the digital picture of the water contact angle (WCA) measurement of PU composites are displayed in Figure 14. The neat PU sample displayed a WCA of 72.87°. The angle was enhanced after the filler’s addition. For the GNP-based composites, the WCA increased from 81.07° to 91.21° upon increasing the concentration of GNP from 0.01 to 0.05 wt%. A similar observation was found for GNR-filled PU composites, where the WCA got higher from 76.14° to 92.38° with increasing content of GNR. The more hydrophobic nature with the addition fillers (GNR or GNP) is expected due to its graphite interlayer structure and uniform dispersion which in terms influences the surface morphology of the composite films.
50
(a) Bar chart, and (b) Digital images of water contact angle of PU composites.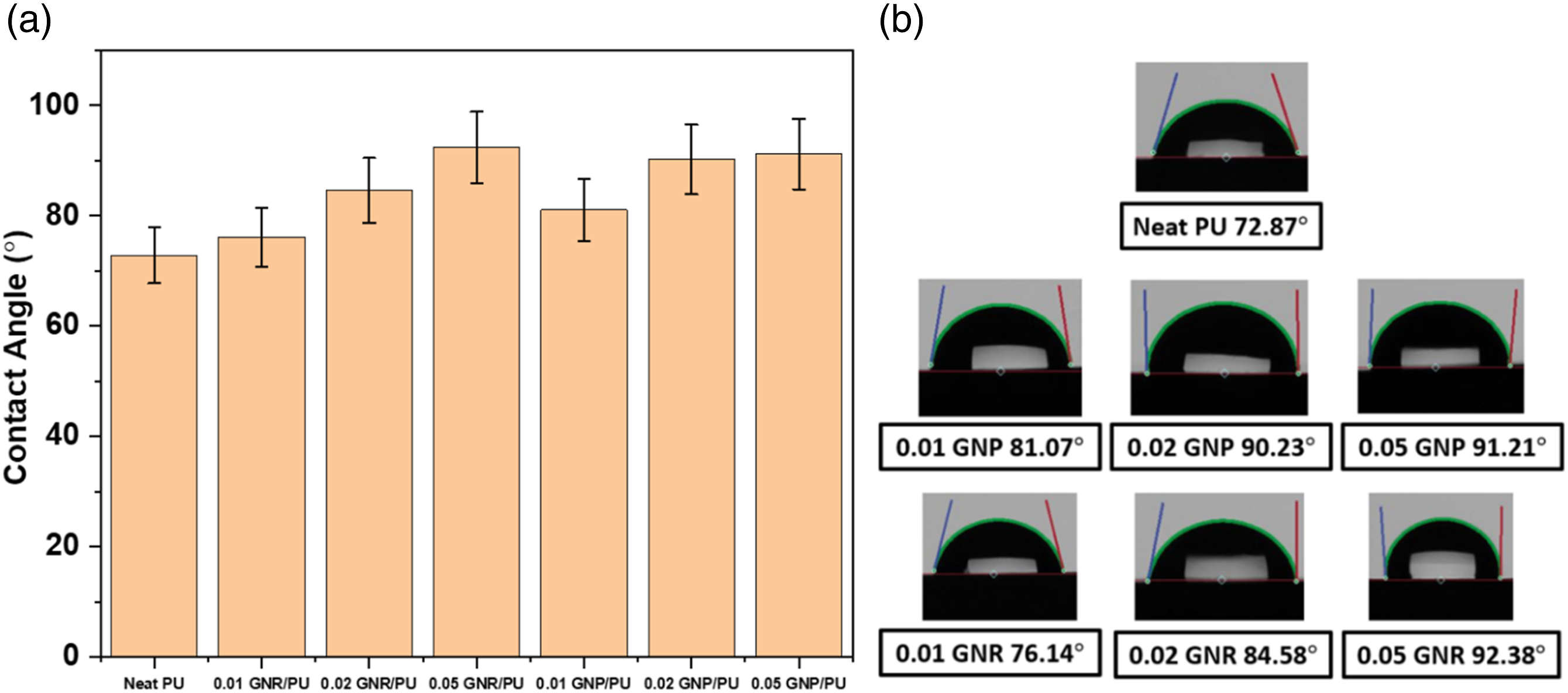
Morphological analysis of the PU composites
The dispersion of graphene derivatives in PU was examined using AFM imaging, as displayed in Figure 15. The analysis was performed in tapping mode to observe the dispersion of GNR and GNP fillers in the PU matrix. Both GNR and GNP fabricated composites showed relatively higher surface roughness than the pure PU film, as obtained from the differences in the height images. This indicates that the graphene fillers are well-distributed within the PU matrix. With increasing content of GNR or GNP, non-uniform dispersion and agglomeration of nanoparticles in the polymer matrix were observed resulting in mild increase in the surface roughness. The addition of fillers like nanoclays,
51
carbon nanotubes,
40
and GO
52
to the PU matrix can cause phase separation in the polymer. AFM images of the PU composites samples with varying GNR and GNP content.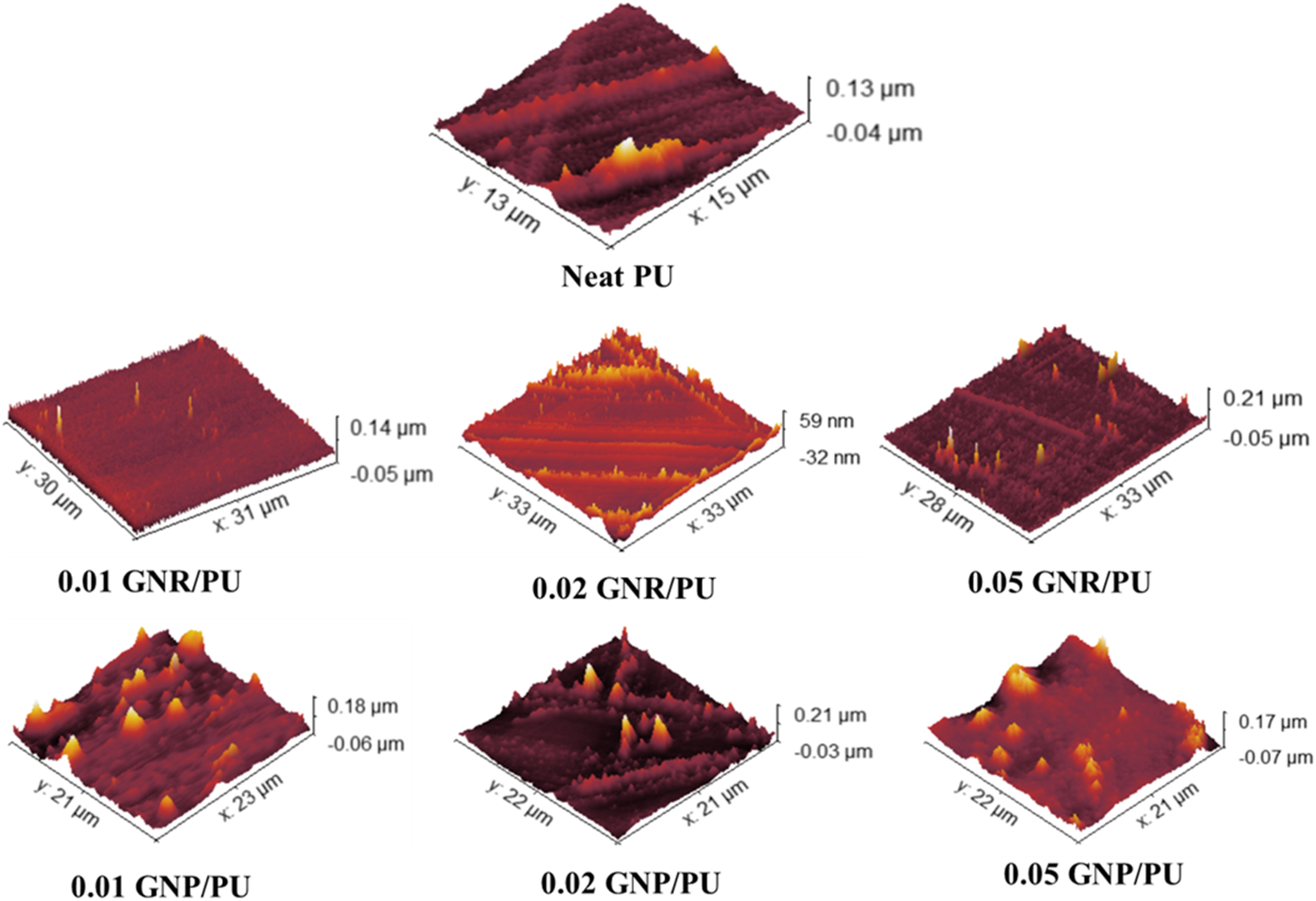
Chemical resistance
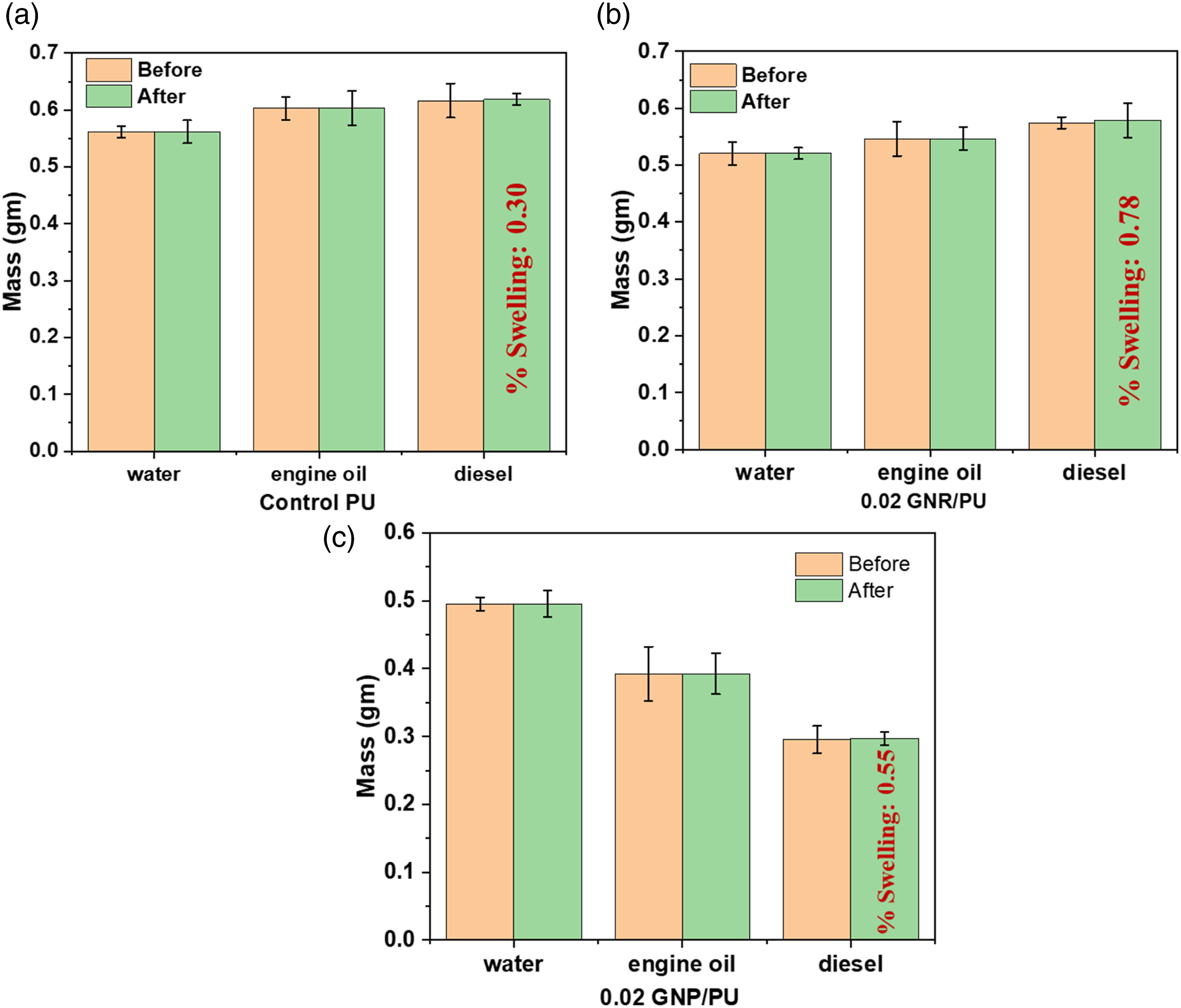
The chemical resistance of the PU, 0.02 wt % GNR, and 0.02 wt % GNP composite films was also investigated (Figure 16). The films were immersed into various chemicals such as water, engine oil, and diesel for 24 h. The pure PU films didn’t show any mass loss or swelling behavior after immersion in all solvents. 0.02 GNR/PU and 0.02 GNP/PU films did not change their form in water, engine oil. The films showed no swelling or mass loss in water and engine oil. However, 0.02 GNR/PU and 0.02 GNP/PU films were slightly swelled in diesel and mass change after 24 h immersion was observed. Therefore, it can be confirmed that the graphene coated composite films displayed good chemical-resistance to solvents. The bar diagram representing the mass change before and after immersion in respective solvents are shown in Figure 17. Pictures of (a) neat PU immersion films, (b) 0.02 wt% GNP/PU and (c) 0.02 wt% GNR/PU films in water (A1), engine oil (A2), and diesel (A3). Bar chart for weight loss differences of (a) neat PU, (b) GNP/PU, and (c) GNR/PU composite films in different solvents.
Conclusion
In summary, the conducted research work efficiently put forward a sustainable use of sunflower oil for polyol synthesis. Epoxidation followed by a ring-opening reaction mechanism was employed for the conversion of SO to polyol. The synthesized polyol was characterized by various techniques, like FTIR, GPC, Viscosity, and analytical determinations. From the FTIR spectra, the appearance of an epoxy-ring vibration peak at 828 cm−1 confirms the formation of ESO, and later the presence of a -OH stretching peak in the polyol proves the successful conversion. From GPC analysis, the synthesized SO-based polyol exhibited a lower retention time of 22.27 min than pure SO at 23.15 min, defining a higher molecular weight of the material. The synthesized polyol was blended with GNP or GNR fillers and isocyanate to fabricate polyurethane composites. The addition of different contents (0.01, 0.02, 0.05 wt %) of graphene derivatives into the PU matrix provides better mechanical strength compared to the pristine polymer. In terms of tensile strength, it was increased from 9 MPa (neat PU) to the highest around 35 MPa for 0.02 wt% GNP loading sample, showing improvement by four folds. For 0.01 wt% GNR-filled composite, the tensile strength obtained was 14 MPa. The flexural strength was enhanced from 3 MPa (neat PU) to 10 MPa by adding 0.01 wt% GNR to the PU matrix. The flexural strength was enhanced by 8 MPa for 0.02 wt% GNP loaded composite films. Shore D hardness was improved from around 53 to 70 Shore D for 0.05 wt% loading of GNP to the PU matrix. There was a slight increment in thermal stability upon adding GNP or GNR as a filler. The composite films also demonstrated hydrophobic nature with increasing filler loadings, as indicated by the WCA analysis. The reinforcement effect of filler in the PU matrix was supported by the enhancement of storage modulus, where the maximum storage modulus for 0.02 wt% GNP/PU and GNR/PU composite films is 1121 MPa and 1487 MPa, and 892 MPa for PU. Morphological properties of the composites supported the formation of higher surface roughness at higher incorporation of GNP or GNR fillers.
Recommendations and future research
Although, this study primarily focuses on the fabrication of a PU composite synthesized from sunflower oil (SFO)-based polyol and the effect of graphene nanofillers (GNPs and GNRs) on the composite performance. The uniform dispersion and efficient interfacial interaction of filler and polymer matrix is a real challenge until today. To improve the interfacial interaction between PU and these inorganic fillers like GNPs and GNRs, surface chemical treatment is a possible way for future scope. Thus, functionalization of graphene fillers can be studied in the future for physico-mechanical properties improvement of composites for multi-faceted applications. The composite film can then be used for further application study like gas barrier properties, making electrically conductive materials and for mobile gas storage applications. As a contribution to further studies, functionalized vegetable oil may come up with interesting results. Thus, we expect that the discussions provided in this part may aid other scientists to incorporate some of the strategies or ideas in their future research.
Footnotes
Acknowledgements
The authors are grateful to the National Institute of Standards and Technology (NIST award number 70NANB20D146) and the U.S. Economic Development Administration (US-EDA award number 05-79-06038) for providing research infrastructure funding.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.