
Abstract
The aim of this research was to study the mechanical and thermal properties of composites based on polylactic acid (PLA) and two types of reinforcements (spent kraft fibers and sisal fibers, with or without modification by silanes). Two different silane coupling agents (3-aminopropyltriethoxysilane and 3-(trimethoxysily)propyl methacrylate) were selected based on previous experiments. Silanization of fibers was measured by ash content, atomic absorption spectroscopy, X-ray photoelectron spectroscopy (XPS), and scanning electron microscopy (SEM) images. PLA-based composites were manufactured by injection molding, and their thermal and mechanical properties were measured by differential scanning calorimetry (DSC) and by tensile and impact tests, respectively. Composites containing kraft and sisal fibers showed remarkable impact strengths (14.45 J/m and 24.61 J/m, respectively), which were 1.4 and 2.4 times higher than the results achieved with neat PLA probes, respectively. Microscopic analysis revealed a good distribution of reinforcements in the PLA matrix.
Introduction
An intensive research is being focused on the development of biobased and biodegradable plastics, fostered by both the depletion of petroleum resources and the increasing awareness in reducing environmental pollution. 1 Polylactic acid (PLA) is the most important biobased thermoplastic polymer. Its precursor, lactic acid, can be easily produced by fermentation of glucose solutions obtained by hydrolysis of renewable feedstocks (containing starch or cellulose), whereas PLA can be produced from lactic acid either by ring-opening polymerization of the lactide or by polycondensation of monomers. 2
PLA shows mechanical properties comparable to petroleum-based plastics and can be processed by conventional methods such as extrusion, injection molding, thermoforming, or compression molding. 1 Natureworks, the major PLA producer, manufactures over 140 000 tons/year 3 and expects to double this amount with a new production plant in China. 4 PLA production entails less energy consumption and lower carbon dioxide emissions than traditional polymers,5,6 and can be biodegraded or composted in a relatively short period of time. However, a wider PLA utilization is hindered by its brittleness, low heat resistance, and comparatively high market price. In this context, PLA reinforcement with a suitable material may result in both enhanced mechanical properties and decreased costs.
Natural fillers (such as starch, hemp, jute, rice, cellulose, kenaf, and wood fibers) have been studied as alternatives to conventional reinforcing materials (glass, aramid, carbon, etc.), due to their low density, annual renewability, biodegradability, low cost, and competitive specific mechanical properties. 7 Sisal fibers, obtained from sisal plant leaves as long fiber bundles, present good mechanical properties compared to other feedstocks7,8 and have been used as reinforcements 9 for thermosets, 10 thermoplastics, 11 or other matrices (for example, chitosan) in the manufacture of food packaging films. 12
However, the major drawback of natural fibers as plastic reinforcements is their limited interaction with the matrix. Load transfer from matrix to fibers is limited by the poor compatibility between the hydrophilic fibers and the hydrophobic polymer matrix. Additionally, fibers tend to agglomerate owing to the high hydroxyl content of cellulose, making a proper distribution into the matrix difficult. In order to improve both compatibility and dispersion, the fiber surface can be subjected to chemical modification to reduce polarity. Additionally, the mechanical and physical properties of composites depend on other factors such as surface roughness, particle aspect ratio, fiber content, and fiber orientation.
Fiber surface modification using organosilanes is an attractive possibility for improving the reinforcement properties. Organosilanes are the major group of coupling agents for glass fiber or mineral-reinforced polymers 13 and are used with virtually any polymer matrix or cement. The surface of natural fibers contains hydroxyl groups, requiring silane transformation into the corresponding silanol for further reaction. Alkoxysilanes undergo hydrolysis leading to the corresponding silanols, which can give polysiloxanes upon condensation, 14 or react with hydroxyl groups at the cellulosic fiber surface. The choice of operational conditions in this stage is very important to ensure the attachment of sylanol to the surface. 14 Silanization can be carried out by either immersing cellulose fibers in an aqueous solution containing silane at an appropriate concentration, or by spraying the fibers with a solution containing suitable reagents.
The amount of water bound to wood fiber is a critical factor in silane reaction.15,16 Insufficient water can lead to the formation of an incomplete monolayer on the fiber surface, whereas an excess of water may result in silane polymerization rather than in reaction with the fiber. Silane concentration is another key factor affecting silanization: Valadez-Gonzalez et al. 17 found that silane binding to henequen fibers decreased when the silane concentration in solution (measured in terms of weight fraction) was above 0.01, due to the preferent formation of polysiloxanes.
Salon et al. 14 found that acidic media favored the hydrolysis reaction and limited the self-condensation of the ensuing silanol groups. In a related work, Bel-Hassen et al. 18 determined that acid catalyzed hydrolysis of silane resulted in the formation of a high amount of silanol groups, reduced the silanol self condensation, and stabilized the proportion of intermediary species for several days. Oppositely, condensation reactions proceed quickly under alkaline conditions, leading to large, three-dimensional structures. Nevertheless, adsorption isotherms of silane onto cellulose showed better results in the case of neutral pH than under acidic conditions, possibly because the silane hydrolysis (giving rise to silanol groups) is not necessary to promote adsorption, promoted by van der Waals interactions rather than by hydrogen bonding.
The terminal functional group of the silane chain plays an important role on silanization. As an example, Abdelmouleh et al.19,20 found that different silane coupling agents (methacrylate, amine, phenyl amine, or mercaptan) resulted in different adsorption isotherms. They concluded that the interaction between fiber and silane were weak and probably driven by hydrogen bonds, enabling the removal of adsorbed silane by Soxhlet extraction. Treatment of silane-containing fibers at 110°C for 2 h resulted in chemical grafting, with 75%–100% silane remaining bound after Soxhlet extraction. Castellano et al. 21 used prehydrolyzed silanes at temperatures about 80°C for chemical modification of the cellulose surface.
Sreekala and Thomas 22 reported 50% decrease in the hydrophilicity of cellulosic fibers upon silane treatments, with little alteration in their mechanical properties. Reduction in hydrophilicity of silane-treated cellulosic fibers (based on water contact angle measurements) was also reported by Abdelmouleh et al. 20 Lu et al. 23 reached similar conclusions for silanized cellulose microfibers, whose crystalline structure was not affected by processing.
Limited effects on the mechanical properties were reported when silane-treated natural fibers were used as reinforcements for thermoplastic matrices. Xie et al. 24 reported modifications in the tensile strength of composites (formulated with LDPE, PS, PP, or PVC as polymeric matrices) ranging from 27% decrease up to 12% improvement, whereas Valadez-González et al.17 achieved 25% increase in the tensile strength of henequen-HDPE composites.
Some studies have been carried out for composites using PLA as a polymeric matrix. Huda et al. 25 reached 17% increase in flexural strength when a silane-treated talc was added to a composite also containing PLA and newspaper fiber. Lee et al. 26 did not find variations in the tensile strength of PLA-talc composites when silane was added in the compounding process. Huda et al. 27 reported an improvement (from 42 to 60 MPa) in the flexural strength of PLA-kenaf fiber composites when the reinforcement was silanized, but both results were below the result achieved for neat PLA (100 MPa). Gregorova et al. 1 determined slight increases in tensile strength of PLA-sitka fiber composites when the reinforcement was treated with silane. Utilization of silane-treated fibers in PLA-kenaf composites resulted in improved tensile and flexural properties and in less water absorption. 28
Different approaches can be employed to assess the influence of silane-containing fibers in the crystallization of PLA. Pei et al. 29 found a significant increase in crystallization rate when using silanized cellulose nanocrystalls, while Wang et al. 30 reported that the nucleation ability of PLA matrix was not affected when sisal fibers were subjected to alkali or silane treatments.
This work provides an assessment on the mechanical and thermal properties of composites made up of PLA and natural fibers (sisal and kraft cellulose fibers) modified by selected silane coupling agents.
Materials and methods
Materials
Spent kraft cellulosic fibers of Eucalyptus globulus wood were obtained from the wastewaters of a local pulping industry (ENCE, Pontevedra, Spain). Samples were defibered using an Ultraturrax laboratory device, filtered, washed with tap water, and air dried.
Sisal fibers were obtained from a rope manufacturer (SICOR, Sociedade Industrial de Cordoaria, S.A., Cortegaça, Ovar, Portugal), treated in a Willey mill, and sieved to select the particles with sizes between 150 and 500 µm.
Coupling agents used in this work
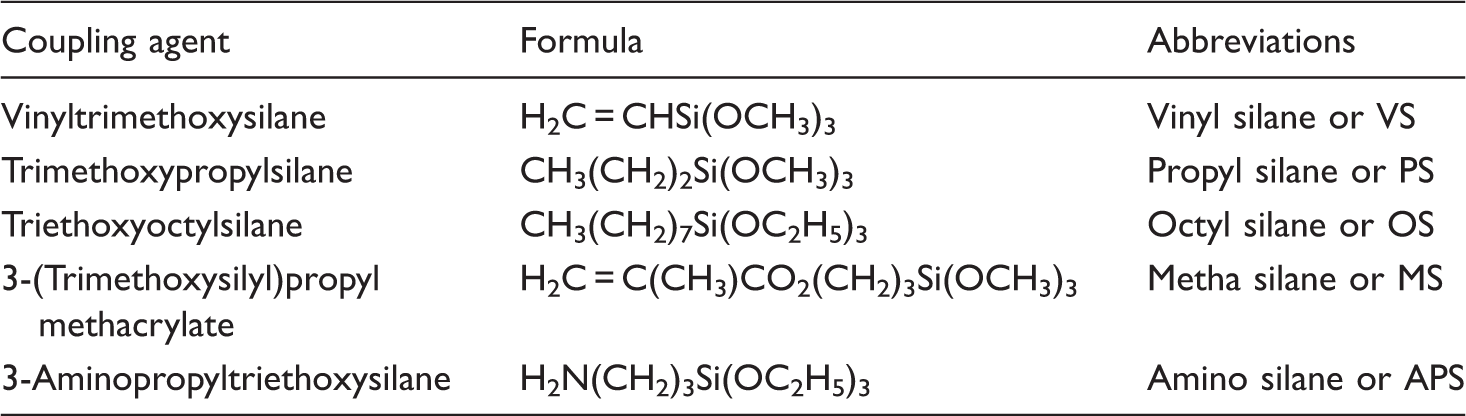
Polylactic acid (PLA) Nature Works 3051 was supplied by Cargill Dow LLC, USA. Its properties were density, 1.25 g/cm3; glass transition temperature (Tg), 59°C; melting temperature (Tm), 152°C; and melt flow index (MFI), 30.3 g/10 min (measured at 210°C and 21.2 N).
Fiber characterization. Aliquots of natural reinforcements were subjected to moisture determination (oven drying at 105°C, method ISO 638), ethanol extractives (TAPPI T-204-cm-97 method), ash (TAPPI T-211-om-93 method), and to quantitative acid hydrolysis (TAPPI T-222-om-98 method). The hydrolysis residue from the quantitative acid hydrolysis was considered as Klason lignin.
The high-performance liquid chromatography (HPLC) analysis of liquors from quantitative acid hydrolysis allowed the determination of glucose (measuring the cellulose content), xylose and arabinose (measuring the hemicellulose content), and acetic acid (coming from acetyl groups bound to hemicelluloses). HPLC analyses were carried out using an Agilent 1200 Series chromatograph equipped with a refractive index detector and an Aminex HPX-87H column (Bio-Rad, CA) under the following conditions: mobile phase, 0.003 mol H2SO4/L; flow rate, 0.6 mL/min; temperature, 50°C. Uronic acids were assayed spectrophotometrically using galacturonic acid as a standard for quantification. 31
Preliminary silanization assays
Preliminary experiments were carried out to assess the comparative suitability of selected silane coupling agents (VS, PS, OS, MS, and APS, see Table 1 for nomenclature) for fiber derivatization. Fibers (10 g) were soaked for 24 h at 25°C into 100 g of acetone with 1 g of the considered silane coupling agent, recovered by filtration, washed with acetone, and dried at room temperature. The silicon content was measured by atomic absorption spectroscopy after solid digestion.
In order to assess the influence of selected operational variables on silanization, additional experiments were carried out using kraft fibers and the best modifier (APS), under conditions defined by different values of the liquor-to-solid ratio (LSR), acetone/water mass ratio, APS loading, pH, and drying temperature (see below). Fibers were immersed for 24 h in acetone-water solutions containing the desired amount of coupling agent, and filtered. Some samples were washed with acetone-water solutions and dried at room temperature, whereas in other cases, wet samples were oven-dried at 100°C for 24 h and washed with acetone-water solutions.
Fiber silanization
Fibers were soaked into a 90:10 w:w acetone:water solution containing 10 wt% of alkoxysilane coupling agent (measured respect to the fiber dry mass) for 24 h. After this time, fibers were slurried and oven-dried at 100°C for 24 h. Dry fibers were filtered and washed with acetone to remove unreacted silane, and air dried. According to ref. 15, the reaction in the surface fiber and the further interaction with PLA can be described as follows: first, silane reacts with water bound to the fiber surface, yielding the corresponding silanol, which reacts with the available hydroxyl groups (in fibers or from other silanol molecules in solution) to form ethers. When silanized fibers are combined with PLA, other functional groups in the silane may react with functional groups of the polymeric matrix. This can be interpreted by the following mechanism:
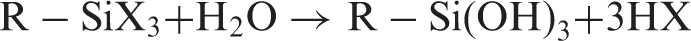
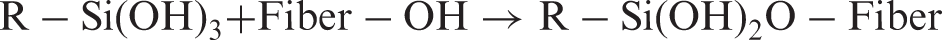

Silicon content of modified fibers
In order to assess the reaction between silanes and cellulose fibers, both untreated and silanized fibers were characterized by different techniques: atomic absorption spectroscopy, increase of ash content, and X-ray photoelectron spectroscopy (XPS).
Atomic absorption spectroscopy. Atomic absorption spectroscopy was performed in a Varian Spectra AA 220/FS. Samples were submitted to acid digestion in a microwave CEM MARSXpress, based on the XprAG-4 method supplied by the microwave manufacturer for wood chips. The digestion method was as follows: 0.25 g of the sample was put into a 55-mL vessel and 5 mL of 70 wt% nitric acid, 0.5 mL of 49 wt% hydrofluoric acid, and 2 mL of 37 wt% hydrochloric acid were added. The vessel was closed, heated up to reach 200°C (heating time, 15 min), kept at this temperature for 15 min and cooled. Then, 15 mL of boric acid was added, and the vessel was closed and submitted to a second heating up to 160°C (heating time, 10 min), kept at this temperature for 10 min, cooled, and the medium was filtered through 45 µm membranes to recover fibers.
Ash content. Ash was determined by sample calcination at 950°C for 3 h. The resulting solid (ash) was treated with hot chlorhidric acid to remove possible metal oxides, filtered, and calcined under the same conditions discussed above. The increase in ash content of silanized samples with respect to unmodified fibers measured the degree of silanization.
X-ray photoelectron spectroscopy. X-ray photoelectron spectra of both untreated and treated fibers were recorded using a Thermo Scientific K-Alpha ESCA instrument equipped with a 1486.6 eV X-ray source. Due to the nonconductor nature of samples, a charge compensation flood gun was employed in experiments. Neutralization of the surface charge was performed using both a low-energy flood gun (0–14 eV) and a low-energy Argon ion gun.
Photoelectrons were collected from a take-off angle of 90° relative to the sample surface. Measurements were done in constant analyser energy (CAE) mode with a 100 eV pass energy for survey spectra and 30 eV pass energy for high-resolution spectra. Binding energies were referenced to the C1s on unsputtered surfaces. Surface elemental composition was determined from the XPS peak areas using the Shirley background subtraction technique and the Scofield sensitivity factors.
Manufacture and properties of composites
Injection molding processing conditions
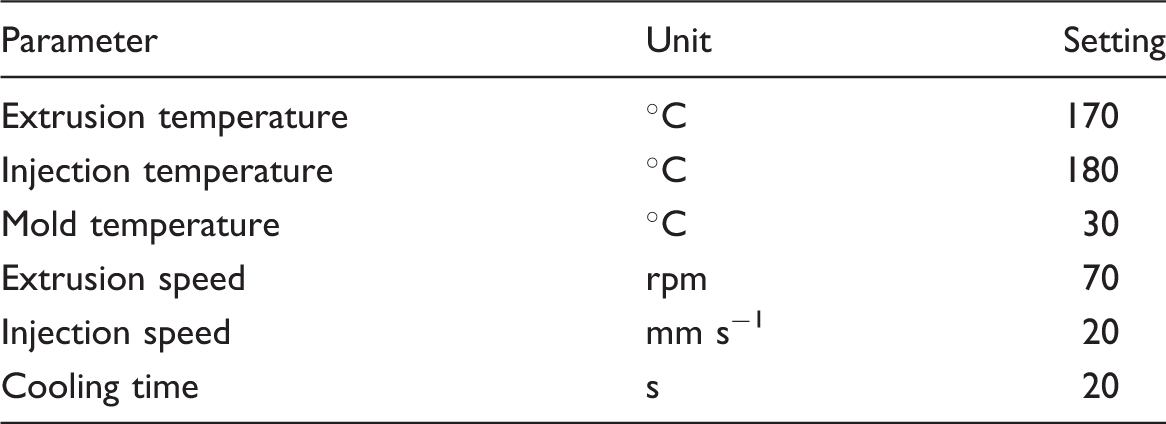
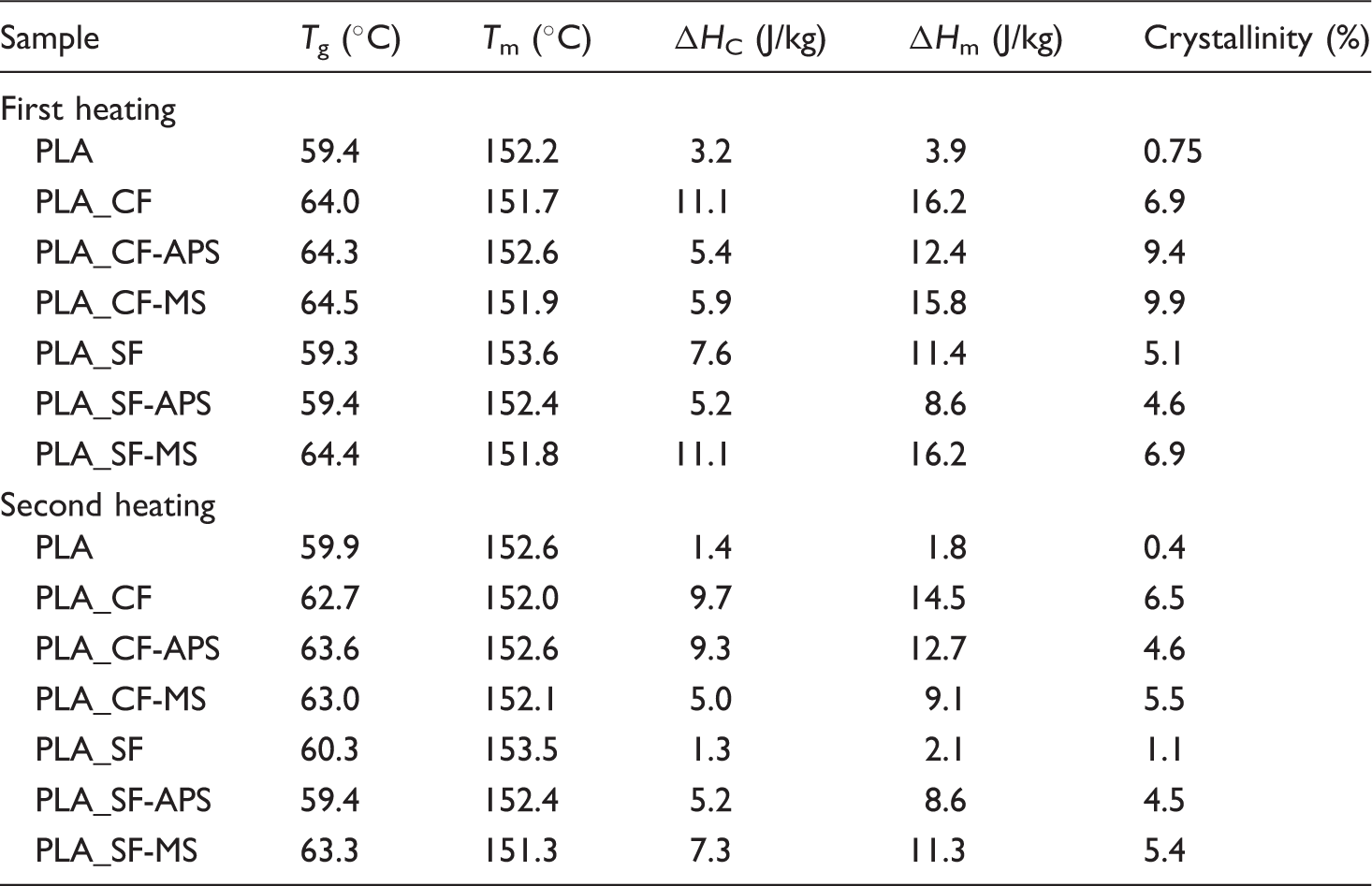
Differential scanning calorimetry (DSC) was used to assess the thermal behavior of composites, defined by the following parameters: glass transition temperature, Tg; crystallization temperature, Tc; pre-melt crystallization enthalpy, ΔHC; melt temperature, Tm; and enthalpy of melting, ΔHm. DSC was performed on a Setaram Setsys Evolution DSC. Samples (20 mg) were heated in DSC pan from room temperature up to 200°C, cooled to room temperature, and heated again up to 200°C. Heating and cooling scans were carried out at a rate of 10°C/min. The percentage of crystallinity (Xc) was calculated using the following equation:
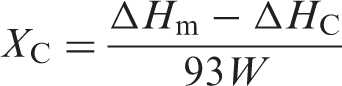
To assess the fiber distribution in the matrix, longitudinal sections of molded specimens were observed using an Olympus BH2 transmission microscope. The observed samples (10 µm thickness) were obtained using a Leitz 40 microtome and mounted on glass lamellae.
Fiber-matrix interfaces were studied by scanning electron microscopy images (SEM) images of selected zones of probe fracture surfaces. Samples were sputter-coated with a thin gold layer, and images were obtained with a Philips XL 30 instrument.
Results and discussion
Material composition
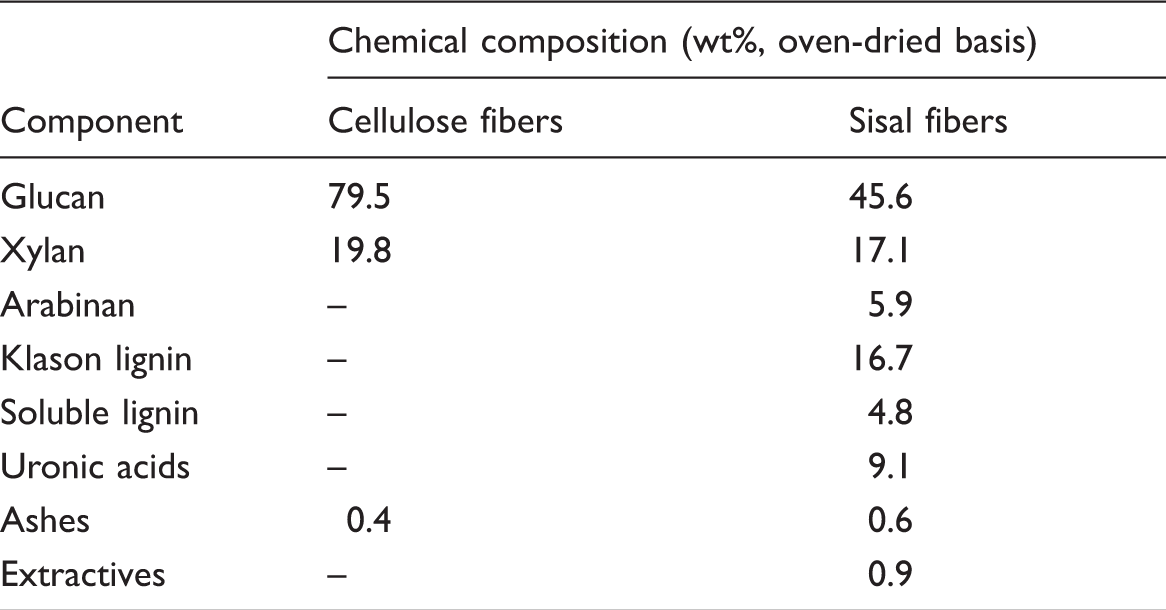
Chemical composition of raw materials
Preliminary silanization treatments
Preliminary experiments showed that APS and MS were bound to the cellulosic fibers upon treatments, while the rest of the assayed silanes presented very low degrees of interaction, with Si concentrations below the sensitivity of the analytical method. The silicon contents of fibers after treatments with APS and MS were 911.8 and 383.3 mg Si/kg sample, respectively. On the basis of these results, both silanes were selected for further experiments.
Experimental conditions in the experiments and silicon concentration in treated samples
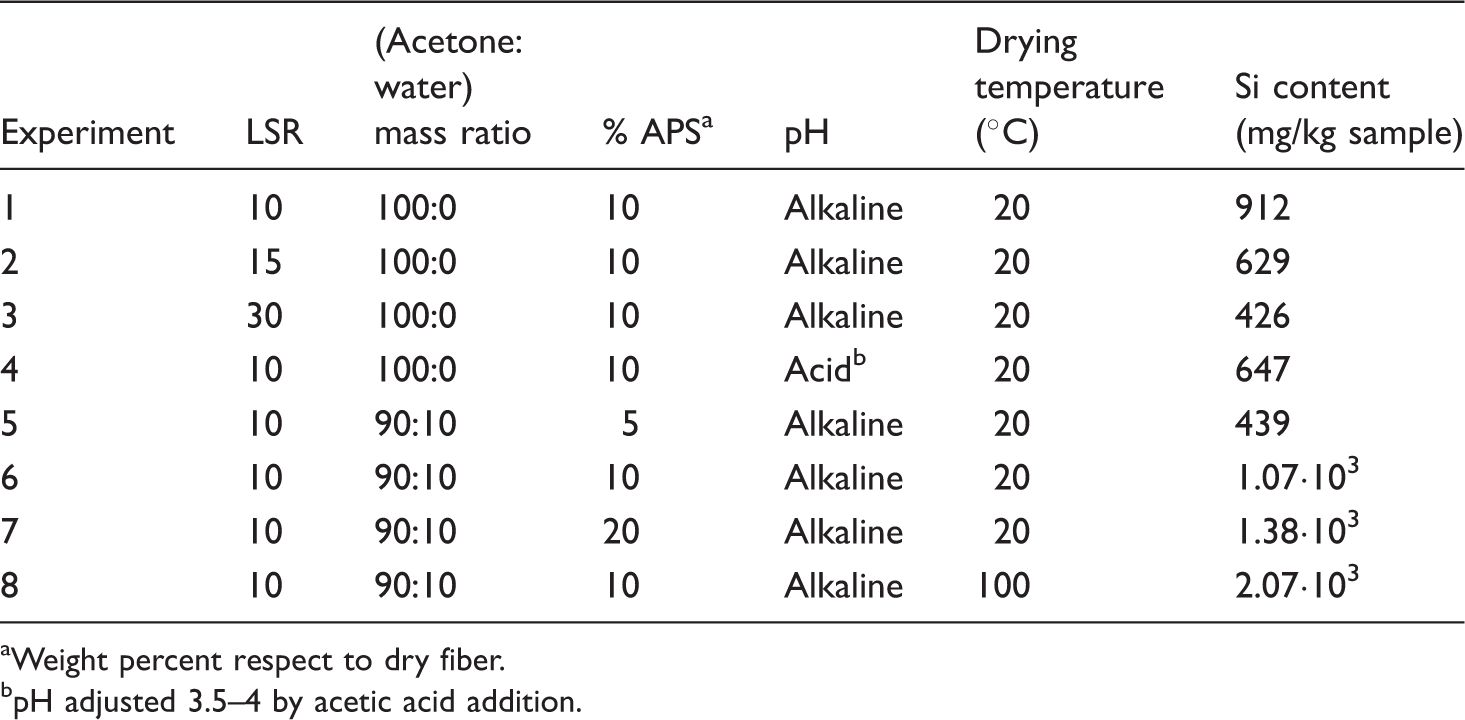
Weight percent respect to dry fiber.
pH adjusted 3.5–4 by acetic acid addition.
In experiments 1–3 of Table 4, the liquor-to-solid ratio (LSR) was modified from 10 (the minimum value ensuring a suitable wetting of solid) up to 30. The best silanization results corresponded to the lowest LSR. In experiment 4, the pH value was kept in the range 3.5–4 by acetic acid addition, and the resulting sample contained less Si than in the case of experiment 1 (performed under alkaline conditions). Experiments 5–7 explored the influence of the silane loading and the effects of acetone: water solutions. Comparing experiments 1 and 6, the highest Si content was obtained operating with acetone-water at 90:10 weight ratio. Considering the effects of the silane loading, a marked improvement was observed in the experiment using 10% instead of 5%, but higher silane loading resulted in minor silanization improvements: for example, doubling the silane loading up to 20% resulted in just 30% increase in the Si content of treated fibers. Oven heating had a remarkable effect on the Si content, resulting in increases of about 100%.
The silicon content of modified fibers measured by atomic absorption spectroscopy showed that the best derivatization conditions corresponded to the experimental conditions of experiment 8, which were selected for surface modification of kraft cellulose fibers and sisal fibers in the preparation of composite reinforcements.
Fiber silanization
Silicon and ash content of samples
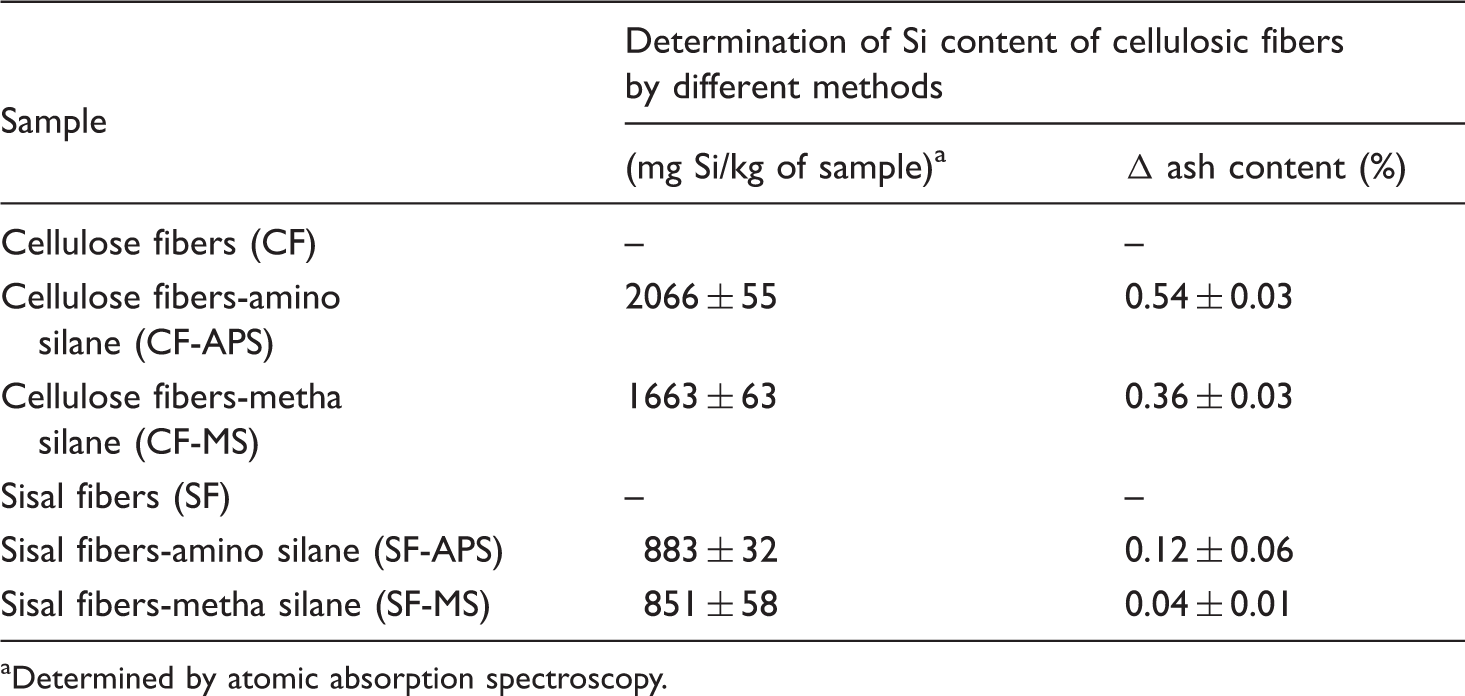
Determined by atomic absorption spectroscopy.
Elemental surface compositions determined by X-ray photoelectron spectroscopy (XPS; as percentages of atom)
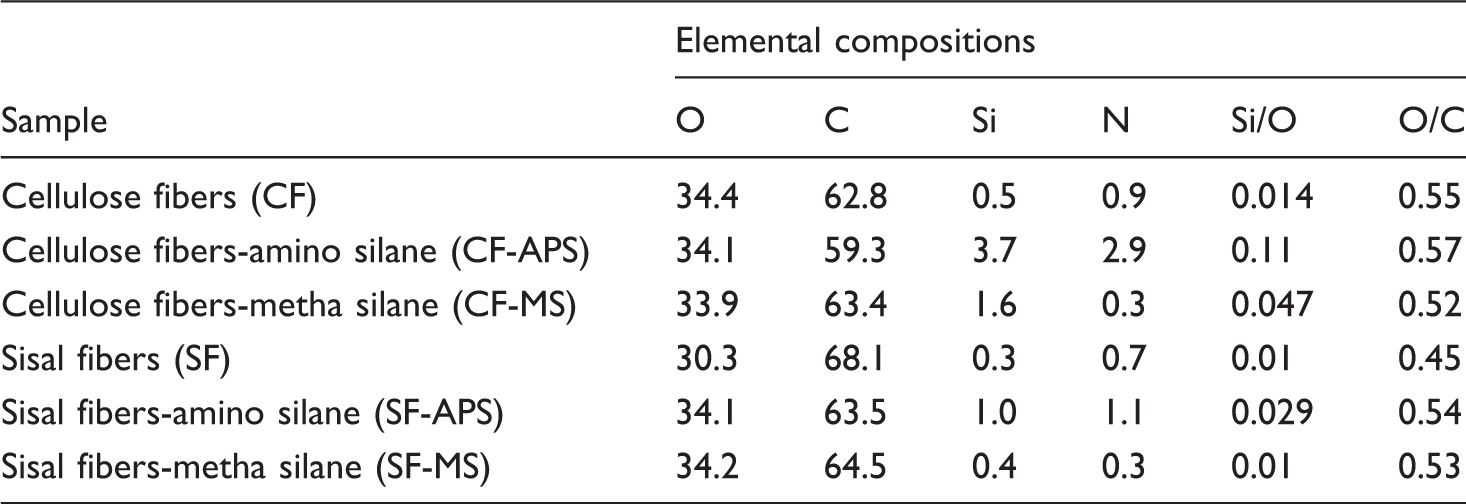
The data in Table 6 highlight the relative amounts of carbon, oxygen, and silicon atoms at the fiber surface. The Si/O ratios were lower than the value of 0.33 calculated for silanization of cellulose, 15 confirming that silanol reacted preferently with hydroxyls present at the fiber surface, with limited occurrence of polycondensation reactions. A pure cellulose sample should present only oxygen and carbon contributions, with an O/C ratio of 0.83. In the case of kraft and sisal fibers, the major peaks corresponded to O and C. The O/C ratio calculated for kraft fibers was close to the ones determined for other cellulosic substrates. 32 In the case of sisal fibers, the lower O/C ratio is due to the presence of lignin, a carbon-rich polymer. Increased N contents were noticed in experiments using aminosilane, in accordance with the chemical nature of this coupling agent.
Analysis of high resolution C1s peaks from XPS spectra
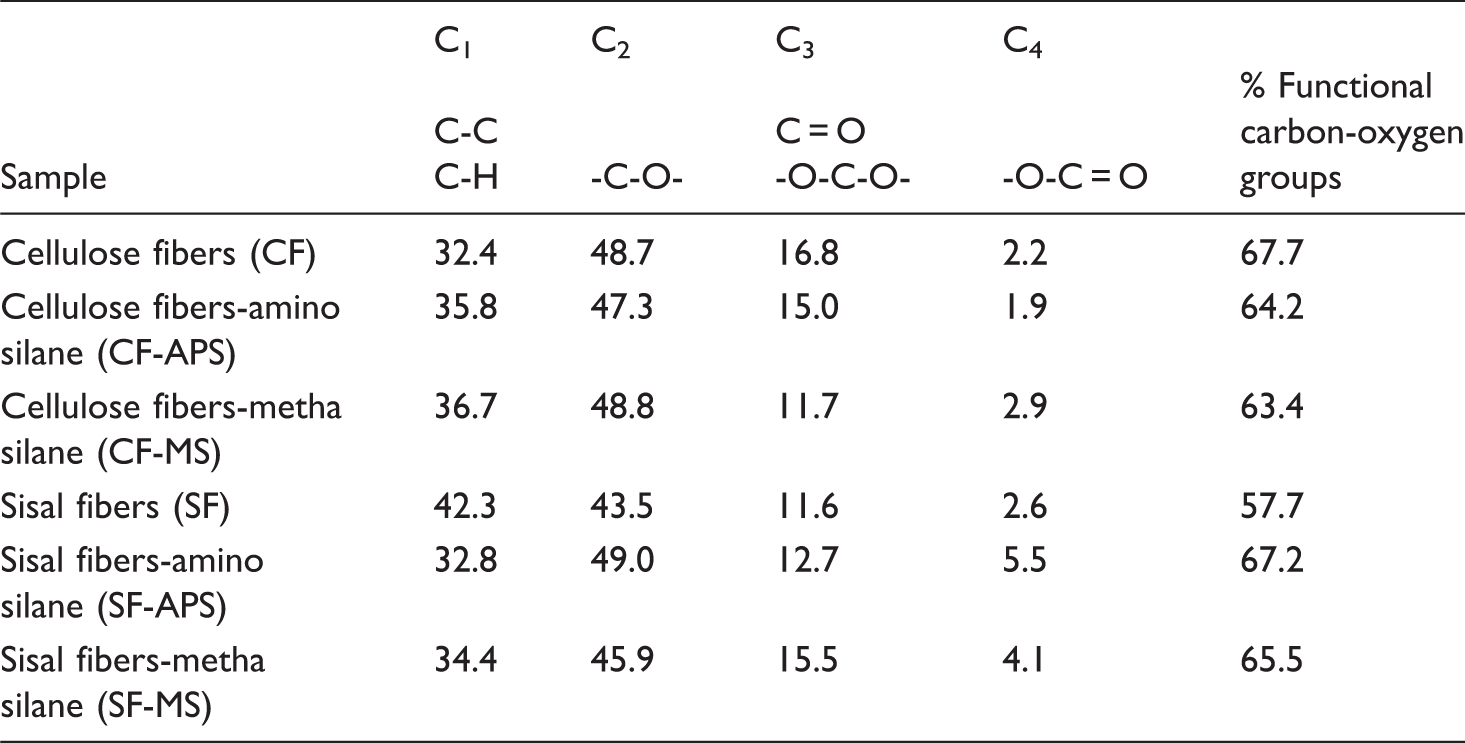
Figure 1 presents the deconvolution of the N1s peak corresponding to CF-APS fibers, resulting in two contributions with bonding energies of 399.50 eV (corresponding to nitrogen in a free amine) and 401.14 eV (corresponding to the protonated ammonium nitrogen).
15
Based on their relative intensities, it can be concluded that 73.9% of nitrogen is in the free amine form. According to Matuana et al.,
15
this indicates that the interaction of silane with fiber surface was mainly achieved by ether bonds between hydroxyls of both silanol and fiber, with minor contributions of interactions between the protonated ammonium and hydroxyl groups at the fiber surface. When APS was used to modify sisal surface, no separation in two peaks was observed.
High resolution N1s spectra of kraft cellulosic fibers treated with amino propyltriethoxy silane (APS).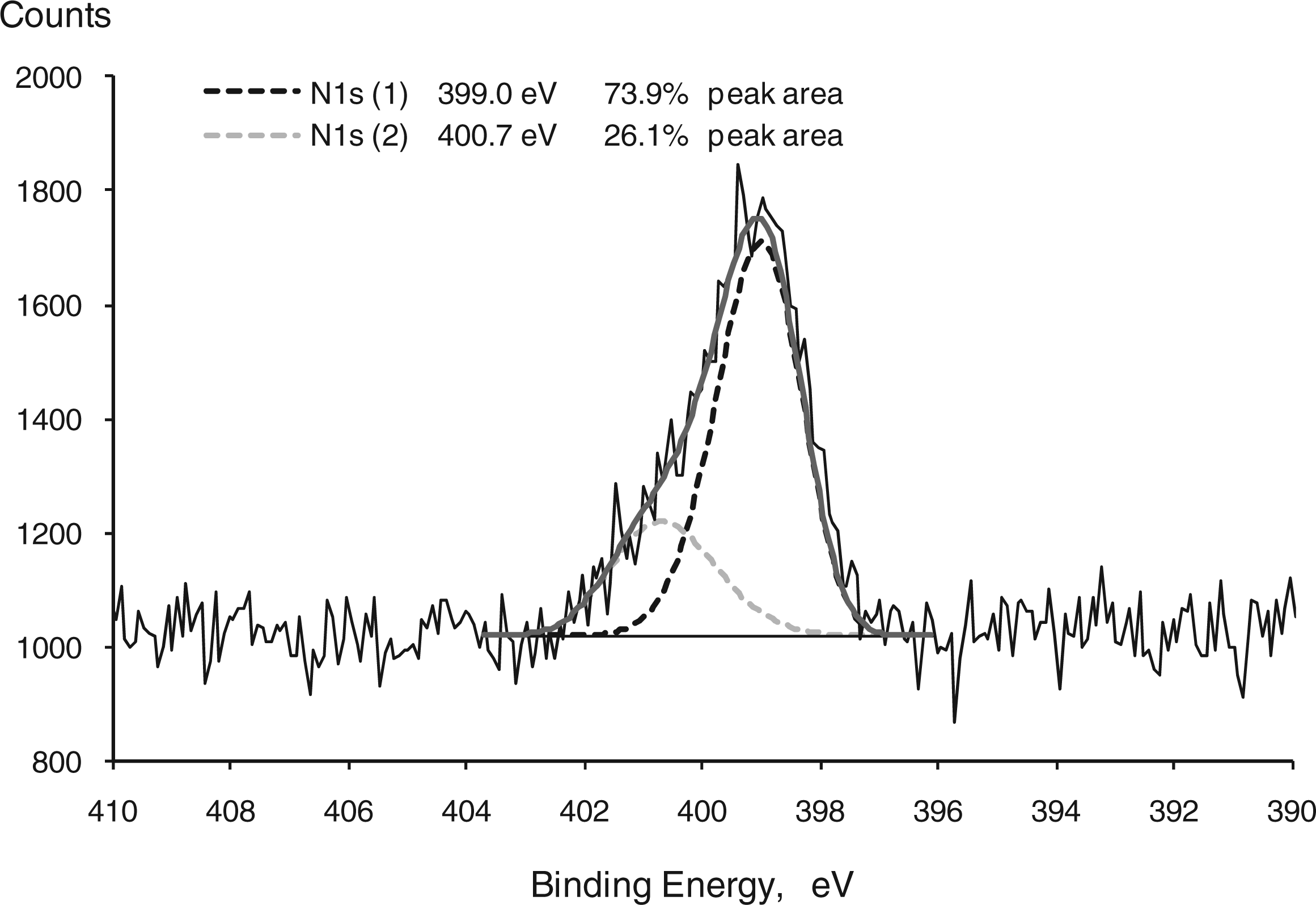
Composite properties
Mechanical properties of composites and pure polylactic acid (PLA) measured by tensile test and Charpy impact test (mean values ± standard deviations)
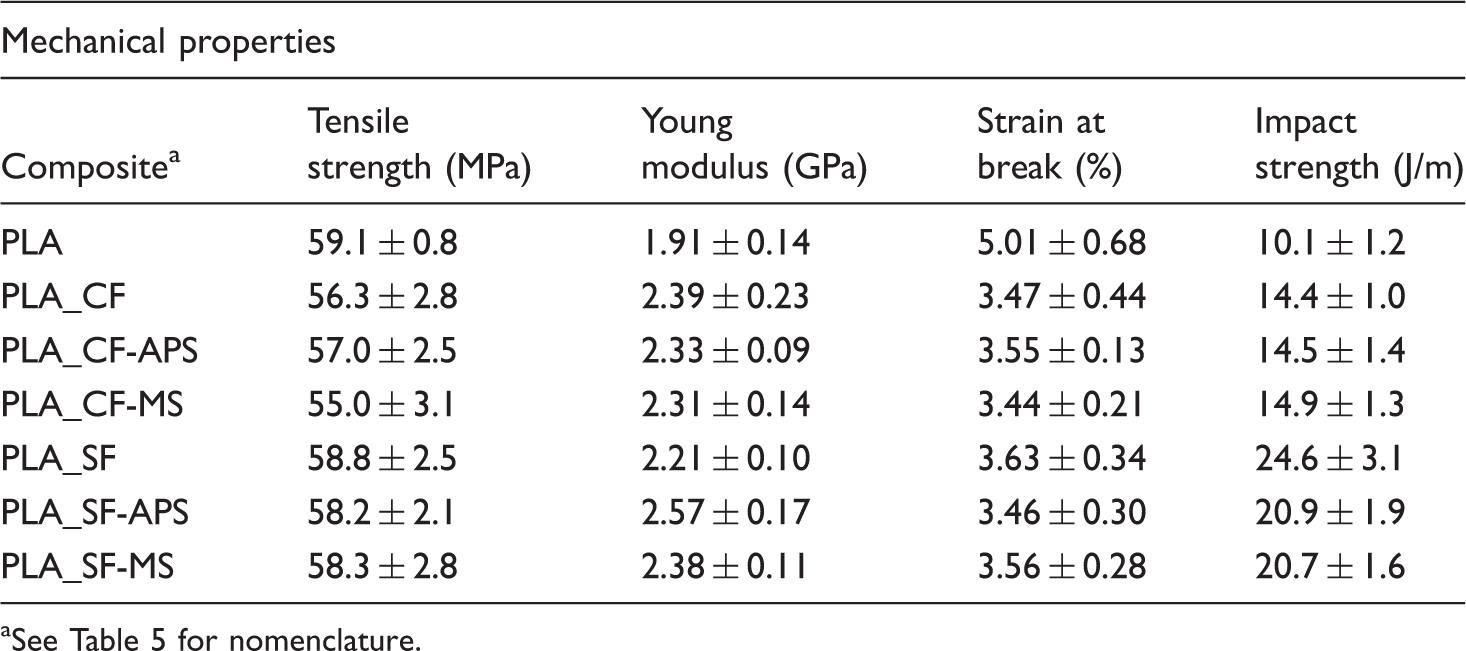
See Table 5 for nomenclature.
Dispersion of the reinforcement materials into the matrix was assessed by optical microscopy of longitudinal probe cuts. Figure 2 shows the experimental situation for cellulose and sisal fibers. The pictures confirmed a good dispersion, with no preferential orientation of fibers.
Fiber distributions for polylactic acid (PLA) composite with sisal fibers (a-b) and kraft fibers (c-d).
Figure 3 shows SEM images of the fracture surfaces of selected probes from tensile tests, where the different size of kraft and sisal fibers can be observed. Kraft fibers presented a quite homogeneous distribution, with some hollows. Different distribution pattern and surface characteristics were observed for APS-treated reinforcements (including kraft and sisal fibers). Sisal fibers perpendicularly oriented with respect to the direction of the tensile force were frequently pulled out.
Scanning electron microscopy (SEM) micrographs (×200) showing the broken surfaces of composite probes from tensile tests. Samples: (a) PLA with 20% cellulose fibers (CF); (b) PLA with 20% cellulose fibers-amino silane (CF-APS); (c) PLA with 20% CF-MS; (d) PLA with 20% sisal fibers (SF); (e) PLA with 20% SF-APS; and (f) PLA with 20% sisal fibers-metha silane (SF-MS).
Thermal properties determined by differential scanning calorimetry (DSC)
Conclusions
3-Aminopropyltriethoxysilane (APS) and 3-(trimethoxysilyl)propyl methacrylate were selected as silanization agents owing to their comparative bonding ability to the cellulosic substrates considered in this work. The highest silanization degree was obtained in treatments using APS at 100°C for 24 h, after soaking fibers in an acetone: water (90:10 wt:wt) solution.
Kraft and sisal fibers (untreated and after silanization) were used as reinforcements for PLA-based composites. The addition of kraft or sisal fibers into the matrix led to composites with improved stiffness and brittleness with respect to PLA, keeping a similar tensile strength. Composites reinforced with kraft and sisal fibers presented remarkable impact strengths (14.45 and 24.61 J/m, respectively), which improved the value determined for PLA probes by factors of 1.42 and 2.43, respectively. Reinforcement did not result in significant changes of Tg and Tm respect to virgin PLA, although the presence of fibers increased the polymer crystallinity. Microscopic analysis revealed a good dispersion of reinforcements in the matrix.
Footnotes
Acknowledgments
Authors are grateful to the Spanish Ministry of Science and Innovation for supporting this study, in the framework of the research Project ‘Properties of new prebiotic food ingredients derived from hemicelluloses’ (reference AGL2008-02072, which was partially funded by the FEDER Program of the European Union). Authors thank Prof. Dr A. M. Cunha and Ms. Sc. A. R. Campos (PIEP, Guimaraes, Portugal) for their support in composite formulation and mechanical characterization, as well as Angel Yanev (PIEP) for technical assistance.