
Abstract
The circadian clock in eukaryotes keeps time via transcriptional feedback loops. In the transcriptional feedback loop of animals, CLOCK activator complexes drive expression of PER repressor complex components which feedback to inhibit CLOCK activation until PER complexes are degraded, thus initiating the next round of CLOCK activation ~24 h later. Recently, we showed that a region of monarch CLOCK (CLK) analogous to that encoded by mammalian CLOCK exon 19 (CLKe19r) and the methyltransferase TRITHORAX (TRX) are required for CLK activation, PER-CLK binding, and PER repression and that TRX-dependent methylation of Heat Shock Protein 68 (HSP68) at arginine 45 (R45) is necessary for PER-CLK binding and PER repression. Given that CLK activation and PER repression complexes in Drosophila are comprised of different core components than in monarchs, we tested whether similar mechanisms are used for CLK activation and PER repression in Drosophila. We found that the CLKe19r, TRX and HSP68 are all required for CLK activation yet only HSP68, but not HSP68 R45 methylation, is required for PER repression in Drosophila. These results reveal a well-conserved CLK activation mechanism and a PER repression mechanism that retains HSP68 function but does not require TRX-dependent methylation.
Keywords
Circadian clocks drive daily rhythms in physiology, metabolism, and behavior via transcriptional feedback loops (TFLs). In animals, CLOCK transcriptional activator complexes (CLOCK-BMAL1 in mammals and CLK-CYC in Drosophila) drive expression of PER repressor complex components (PER-CRY2 in mammals and PER-TIM in Drosophila), which feedback to inhibit CLOCK activators until they are degraded to initiate the next cycle of transcription (Lowrey and Takahashi, 2011; Hardin and Panda, 2013). Although the Drosophila clock uses CLK-CYC (hereafter dCLK-CYC) to activate and PER-TIM (hereafter dPER-TIM) to repress transcription, monarch butterflies (Danaus plexippus) possess mammalian-like CLK-BMAL1 (hereafter dpCLK-BMAL1) activators and PER-CRY2 (hereafter dpPER-CRY2) repressors (Yuan et al., 2007; Zhu et al., 2008; Merlin et al., 2013), indicating that the Drosophila clock is derived from an ancestral mammalian-like clock but lacks both CRY2 and the BMAL1 activation domain targeted by CRY2 to repress transcription (Zhang et al., 2017).
In mammals, the histone methyltransferase MLL1 binds to a region of CLOCK encoded by exon 19 to activate transcription (Katada and Sassone-Corsi, 2010). This interaction is conserved in the monarch clock; the MLL1 ortholog TRITHORAX (TRX) binds the conserved exon 19 region of dpCLK (dpCLKe19r) to activate transcription (Zhang et al., 2022). Surprisingly, TRX is not only required for dpCLK activation in monarchs but also for dpPER-CLK binding and dpPER repression (Zhang et al., 2022). Although TRX likely methylates histones to promote dpCLK-BMAL1 activation in monarchs as it does in mammals (Katada and Sassone-Corsi, 2010), TRX also promotes dpPER repression by directly or indirectly methylating Heat Shock Protein 68 (HSP68) at arginine 45, an event required for dpPER-CLK binding (Zhang et al., 2022). Given that dCYC lacks the C-terminal activation domain of BMAL1 and dPER partners with TIM rather than CRY2, we wanted to determine whether the roles TRX, dCLKe19r, and HSP68 play in transcriptional activation and repression in the monarch TFL are conserved in Drosophila.
Previous work in Drosophila showed that the dCLKe19r is important for high levels of dCLK-CYC transcription in vivo (Lee et al., 2016). Here, we confirm that dCLKe19r contributes to dCLK activation and also show that TRX functions as it does in monarchs to promote dCLK-CYC transcription. However, in contrast to monarchs, dPER is still able to bind dCLK lacking the e19r (albeit at low levels) to repress dCLK-CYC transcription, likely due to dPER binding dCLK outside the dCLKe19r (Lee et al., 2016). TRX is not required for dPER repression as it is in monarchs, consistent with dPER repression in the absence of dCLKe19r where TRX can bind. HSP68 is required for CLK activation, CLK-PER binding, and PER repression in both monarchs and flies. However, in monarchs, dpPER-CLK binding and dpPER repression require TRX-dependent methylation of HSP68 at arginine 45, whereas such HSP68 methylation is not required for dPER-CLK binding and dPER repression in Drosophila. These results highlight a conserved role of HSP68 in CLK activation, CLK-PER binding, and PER repression between Drosophila and a mammalian-like clock bearing insect, and suggest that while TRX is integrally involved in PER-CLK binding and PER repression via HSP68 in monarchs, TRX plays little to no role in PER repression in Drosophila.
Methods
Plasmids for Expression in S2 Cells
The pAC-dPer_V5, dPer_Luc, and pACT-Renilla-Luc plasmids were provided by Michael Rosbash (Nawathean and Rosbash, 2004). Construction of all other plasmids for expression in Drosophila Schneider 2 (S2) cells are listed in Supplementary Table S1.
S2 Cell Culture, Transfections, Transcriptional Assays, and Western Blotting
Drosophila S2 cells were maintained at 25 °C in Schneider’s Drosophila medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco) and 100 U/mL penicillin and streptomycin (Gibco). Transient transfections were performed as previously described (Chang and Reppert, 2003) in 12-well plates using 10 ng/well of dPer-Luc as reporter and 30 ng/well pACT-Renilla-Luc as normalization control. S2 cells were co-transfected with 5 ng/well of pAc plasmids expressing wild-type dCLK, mutant dCLK variants, or single domains of dCLK fused to a Gal4 DNA-binding domain (DBD) or a VP16 transactivation domain (see plasmids section above) with or without varying amounts of either pAC-dPer_V5 or pAC_3xFLAG_dPer plasmids. Since S2 cells endogenously express cyc, dCLK engages with endogenous CYC to activate dPer-Luc expression (Darlington et al., 1998). Plasmids were mixed with 5 μL Cellfectin II (Invitrogen), incubated for 15 min at room temperature, and mixed with serum-free Schneider’s Drosophila medium before being added to S2 cells. An equal volume of Schneider’s Drosophila medium supplemented with 20% heat-inactivated FBS and 2% penicillin and streptomycin were added after approximately 6 h of incubation at 25 °C. Cells were incubated for 2 days before harvesting for luciferase assay, quantification of gene expression, and Western blot analysis.
For luciferase assay and Western blot analysis, cells were lysed with 50 μL of 1× Passive lysis buffer (Promega). Firefly and renilla luciferase activities were quantified with a Dual-Luciferase reporter assay system (Promega) using 5 μL of cell protein lysate with 50 μL of Luciferase Assay Reagent II for firefly luciferase activity and 50 μL of Stop & Go Reagent for renilla luciferase on a VICTOR3V Multilabel Plate Counter (PerkinElmer).
For RNA interference (RNAi) of endogenous D. melanogaster (d)Trx in S2 cells, a 430-bp and a 477-bp fragment corresponding to the 5′- and 3′-UTRs of dTrx, respectively, were amplified from S2 cell cDNA using Phusion Taq polymerase (NEB) with the following primers containing a T7 promoter (5′UTR dTrx F: 5′-TAATACGACTCACTATAGGGTGAGTTTTCCACAAGTTCG-3′; 5′UTR dTrx R: 5′-TAATACGACTCACTATAGGGGCCGTATTGCCACTAATTG-3′; 3′UTR dTrx F: 5′-TAATACGACTCACTATAGGGCGTCGAAATTAATCCGTATACTC-3′; 3′UTR dTrx R: 5′-TAATACGACTCACTATAGGGGGTTGTAAAACGAAAAGTATCAC-3′). For RNAi of endogenous dHsp68 in S2 cells, a 444-bp and a 416-bp fragment corresponding to the 5′- and 3′-UTRs of dHsp68, respectively, were amplified from S2 cell cDNA using Phusion Taq polymerase (NEB) with the following primers containing a T7 promoter (5′UTR dHsp68F: 5′-TAATACGACTCACTATAGGGCTACATTTGAAATCAAACAG-3′; 5′UTR dHsp68 R: 5′-TAATACGACTCACTATAGGGCATCGAACACAGAGTTC-3′; 3′UTR dHsp68F: 5′-TAATACGACTCACTATAGGGCAACTGGAGACCTATTTG-3′; 3′ UTR dHsp68 R: 5′-TAATACGACTCACTATAGGGATTTAATAGCATATTGGAAC-3′).
For each target, purified amplicons were in vitro transcribed using OPTIZYME T7 RNA polymerase (Fisher), treated with RQ1 Dnase (Promega), and the 2 single-stranded RNA strands were purified individually by acid phenol-chloroform extraction and annealed together by incubation at 65 °C for 30 min followed by slow cooling to room temperature. Double-stranded RNA of enhanced green fluorescent protein (egfp), used as a control, was generated as previously described (Hung et al., 2007). For transfections of dsRNA-treated cells, S2 cells were treated with a mix of 7.5 μg of dsRNA against the 5′-UTR and 7.5 μg of dsRNA against the 3′-UTR of either dTrx or dHsp68, and other expression plasmids in 500 μL of serum-free Schneider’s medium for 6 h at 25 °C and incubated for 2 days in supplemented medium, as described above. Cells were washed with 1× PBS, harvested by scraping, resuspended in 1× PBS, and divided in 2 aliquots of equal volume for luciferase assay and RT-qPCR, respectively.
For Western blotting, triplicate S2 cell protein extracts lysed with 1× passive lysis buffer (Promega) were combined in equal volume and protein concentrations were measured using Pierce Coomassie Plus Assay kit (Thermo Scientific) and Bovine Serum Albumin as a standard on a VICTOR3V Multilabel Plate Counter (PerkinElmer). For each sample, 10 to 20 μg of equal amount of protein were loaded onto a 7.5% SDS PAGE. V5-tagged dPER was detected using a mouse anti-V5 monoclonal primary antibody (Novex R96025; 1:5000) and FLAG-tagged dPER was detected using a mouse anti-FLAG monoclonal primary antibody (Sigma F3165; 1:2000). In both cases, a goat anti-mouse IgG HRP was used as the secondary antibody (Biorad 170-6516; 1:10,000). Drosophila actin was detected using a mouse anti-beta actin monoclonal antibody (Abcam 8224; 1:5000) and a goat anti-mouse IgG HRP (Biorad 170-6516; 1:10,000). Horseradish peroxidase activity was detected with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific).
Real-Time RT-qPCR
To quantify the levels of endogenous dTrx mRNA in RNAi experiments in S2 cells, cells were lysed in 100 mM Tris-HCl pH 7.0, 100 mM LiCl, 20 mM DTT, and 10% SDS; total RNA was extracted by 2 rounds of acid phenol-chloroform (Ambion) and treated with RQ1 DNase (Promega); and random hexamers (Promega) were used to prime reverse transcription with Superscript II Reverse Transcriptase (Invitrogen), all according to the manufacturers’ instructions. Quantifications of gene expression were performed on a QuantStudio 6 Flex Real-Time PCR System (Thermo Scientific) using iTaq Universal SYBR Green Supermix (BioRad) and the following primers: dTrxF, 5′-TGCAGTCGTCTGTATTGGTCTTC-3′, and dTrxR, 5′-CAGTATAATTTCGACCCACATCCA-3′ for dTrx, and dRp49F, 5′-GCTAAGCTGTCGCACAAATGG-3′ and dRp49R, 5′-CGGCGACGCACTCTGTT-3′ for the Rp49 control. The near 100% efficiency of each primer set was validated by determining the slope of Ct versus dilution plot on a 3 × 104 dilution series. Individual reactions were used to quantify each RNA level in a given cDNA sample, and the average Ct from duplicated reactions within the same run was used for quantification. The data for each gene in any given condition were normalized to rp49 as an internal control and normalized to the mean of one sample within a set for quantification.
Co-Immunoprecipitations
Plasmids and/or dsRNA against dTrx or dHsp68 (amounts given in figure legends) were transfected in S2 cells cultured in 6-well plates with 10 μL of Cellfectin II (Invitrogen). Cells were harvested 48 hours after transfection, lysed on ice for 10 min in 300 μL of extraction buffer (20 mM HEPES at pH 7.5, 100 mM NaCl, 0.5% NP-40, 1× Complete Protease Inhibitor; Pierce), and after centrifugation 50 μL was retained as input sample. For co-immunoprecipitations (IPs) of FLAG-tagged proteins, protein G plus/protein A agarose beads (50 μL; EMD Millipore) were prepared for IP by 3 consecutive washes in 1 mL of extraction buffer. The beads were then incubated with a mouse anti-FLAG monoclonal antibody (5 μL, Sigma F1804) for 1 h at room temperature. The unbound antibodies were removed by an additional wash with 300 μL of extraction buffer. Protein extracts were added to the washed beads and incubated overnight at 4 °C on a rotator. For co-IPs of V5-tagged proteins, anti-V5 agarose (20 μL; Sigma A7345) was prepared for IP by 3 consecutive washes with 300 μL of RBS. Protein extracts were added to the RBS washed beads and incubated 1 h at room temperature and overnight at 4 °C on a rotator. Protein G plus/protein A agarose beads were then washed 3 times with ice-cold extraction buffer and anti-V5 agarose beads were washed 3 times with ice-cold RBS before proteins were eluted by adding 40 μL of 5× protein sample SDS buffer and boiled at 95 °C for 6 min. Complexes were resolved on a 6% SDS-PAGE, and proteins were detected by immunoblotting with the following antibodies: mouse anti-FLAG monoclonal primary antibody (Sigma F3165; 1:2000), mouse anti-V5 monoclonal primary antibody (Novex R96025; 1:5000), and goat anti-mouse-HRP secondary antibody (Biorad 170-6516; 1:10,000).
Statistical Analysis
p-values were calculated using 1-way ANOVAs using the online calculator from http://vassarstats.net, the QI Macros Statistical software for Excel, and the R package.
Results
The dCLKe19r and TRX Are Required for dCLK Activation but Not dPER Repression
The mouse ClockΔ19 mutant eliminates a 51 amino acid region encoded by exon 19 that lengthens and/or abolishes activity rhythms during constant darkness (Vitaterna et al., 1994; King et al., 1997). Mouse clockΔ19 is required for CLOCK-BMAL1-dependent transcription as CLOCKΔ19 protein acts as a dominant negative; CLOCKΔ19 binds BMAL1 and CLOCKΔ19-BMAL1 heterodimers bind E-box regulatory elements but are unable to activate transcription (Gekakis et al., 1998). In monarchs, we previously showed that removing the conserved dpCLKe19r from dpCLK greatly decreases dpCLK-BMAL1 transcriptional activation and eliminates dpPER-CLK binding and dpPER repression (Zhang et al., 2022). Previous results in Drosophila show that a deletion mutant that eliminates the dCLKe19r also compromises transcriptional activation (Lee et al., 2016). Given that the PER repression complex is different in Drosophila compared to monarchs and mammals (i.e. dPER engages with TIM rather than CRY2), we tested whether loss of the dCLKe19r also impaired dPER repression.
Consistent with previous results, activation assays in Drosophila Scheider 2 (S2) cells shows that a dCLK mutant lacking the e19r (dClkΔ19r) reduces transcription of the Per-Luc reporter by ~90% (Figure 1a). Since dClkΔ19r drives such low levels of Per-Luc activation, it was difficult to determine the extent to which PER represses transcription (Figure 1a), so we fused a VP16 activation domain to dCLK and dClkΔ19r to boost the level of activation. With the increased activation by dCLK_VP16 and dClkΔ19r_VP16, we found that dPER repressed both dCLK_VP16 and dClkΔ19r_VP16 driven dPer-Luc expression to a similar extent (Figure 1a). Thus, in contrast to monarchs, these results suggest that dCLKe19r has little impact on dPER repression. We previously showed that expressing increasing amounts of dpCLKe19r with wild-type dpCLK and dpPER almost completely abolished dpPER repression, indicating that dpPER repression is mediated through dpCLKe19r (Zhang et al., 2022). When dCLKe19r was expressed in increasing amounts up to 80-fold excess over wild-type dCLK, the level of dPER repression decreased only ~2-fold (Figure 1b). This result indicates that the dCLKe19r has a negligible impact on dPER repression, consistent with our dPER repression results with dCLK_VP16 and dClkΔ19r_VP16 (Figure 1a).
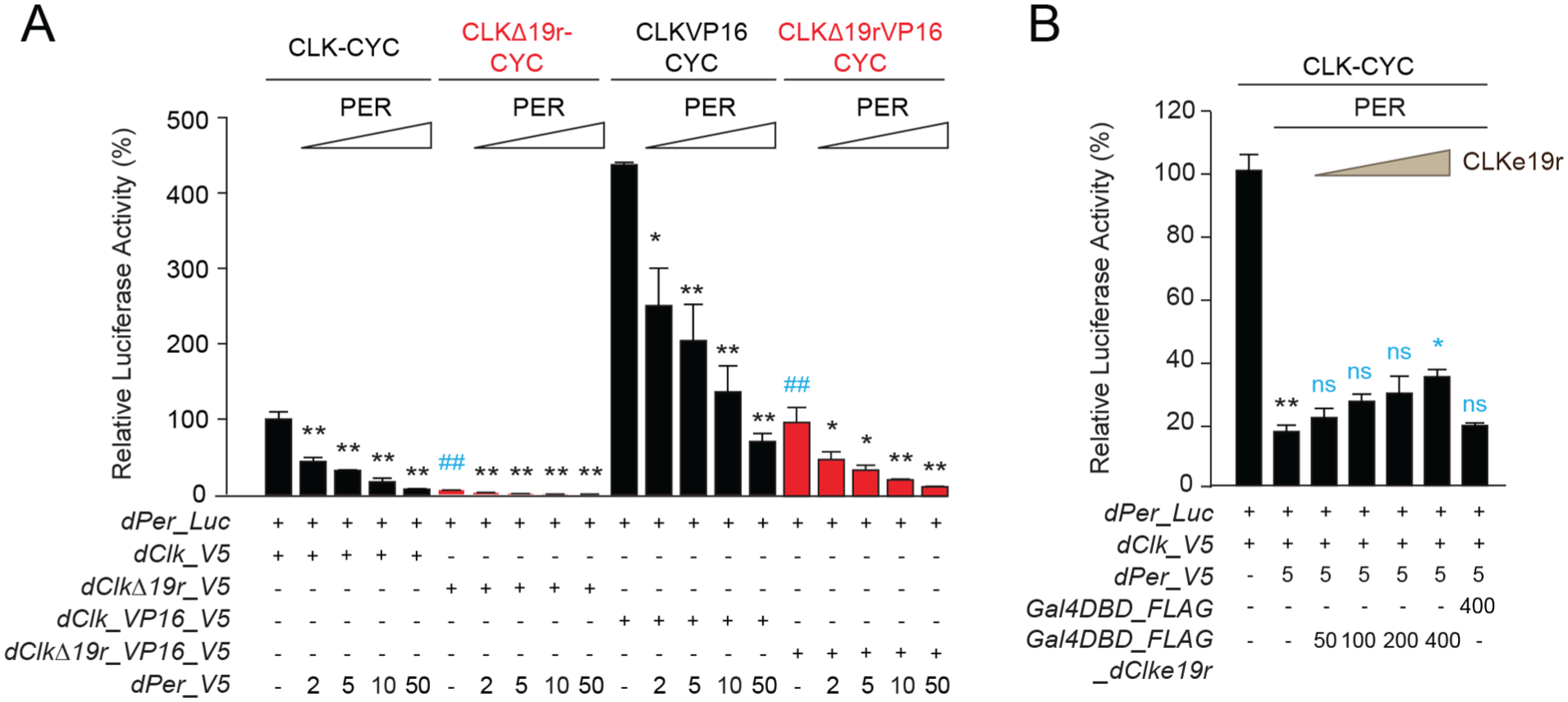
CLKe19r is necessary for CLK:CYCLE-mediated transcriptional activation, but not PER repression. (a) The Drosophila per promoter-driven luciferase reporter (dPer_Luc; 10 ng) was expressed in Drosophila S2 cells in the presence of dCLK, dCLKΔ19r, dCLK_VP16, or dCLKΔ19r_VP16 (5 ng each), with increasing doses of dPER (amount given in ng). Firefly luciferase activity was computed relative to renilla luciferase (30 ng) activity. Each value is mean ± SEM of 3 replicates. For each condition, 1-way ANOVA, Tukey post hoc: ##p < 0.01 (in gray for activation); *p < 0.05, **p < 0.01 (in black for repression). (b) dPer_Luc (10 ng) was used in the presence of dCLK and dPER (5 ng each) in the presence or absence of Gal4DBD or Gal4DBD_dCLKe19r (amounts given in ng). Quantification of luciferase activity and statistics were performed as in (a). One-way ANOVA, Tukey post hoc: **p < 0.01, *p < 0.05, and ns is nonsignificant. The black stars denote repression by dPER and the effects of increasing doses of Gal4DBD_dCLKe19r or a high dose of Gal4DBD on dPER repression are shown in gray.
The histone methyltransferase MLL1 binds the mouse CLOCKe19r and deposits H3K4me3 marks at clock target gene promoters to promote transcription (Katada and Sassone-Corsi, 2010). Likewise, we found that the Drosophila MLL1 ortholog TRX binds dpCLKe19r and activates transcription of a dpPer-Luc reporter, indicating that MLL1/TRX plays a conserved role in activating dpCLK-BMAL1 transcription (Zhang et al., 2022). In addition, we showed that TRX, and in particular TRX methyltransferase activity, is also required for dpPER transcriptional repression (Zhang et al., 2022).
To determine whether TRX contributes to dCLK activation and/or dPER repression, we used RNA interference (RNAi) to knock down Trx expression in S2 cells and measured the levels of dPer-Luc reporter gene expression. In cells expressing Trx RNAi, the level of dCLK activation was decreased by about 30% compared to control cells expressing GFP RNAi and ~60% compared to the no RNAi control (Figure 2a), indicating that TRX contributes to dCLK activation. In addition, dPER repression persists in cells expressing Trx RNAi but was ~2-fold lower than the GFP RNAi and no RNAi controls (Figure 2a). RNAi targeting Trx was effective, reducing Trx mRNA levels by ~70% (Figure 2b). Although dPER effectively represses dPer-Luc transcription independent of the dCLKe19r in S2 cells (Figure 1), whether dPER also represses transcription via the dCLKe19r and TRX was less clear. To test this, we fused the dCLKe19r to a Gal4 DNA-binding domain linked to the VP16 activation domain (Gal4DBD-dClke19r-VP16) and tested whether dPER could repress UAS-driven luciferase (UAS-Luc) in the presence or absence of TRX compared to Gal4DBD-VP16 controls. Our results showed that dPER is unable to repress transcription through dCLKe19r alone in the presence of TRX or when Trx levels are reduced ~60% by RNAi (Figure 2c and 2d). These results show that although dCLKe19r and TRX are both required for dCLK activation, neither dCLKe19r nor TRX contribute substantially to transcriptional repression by dPER.
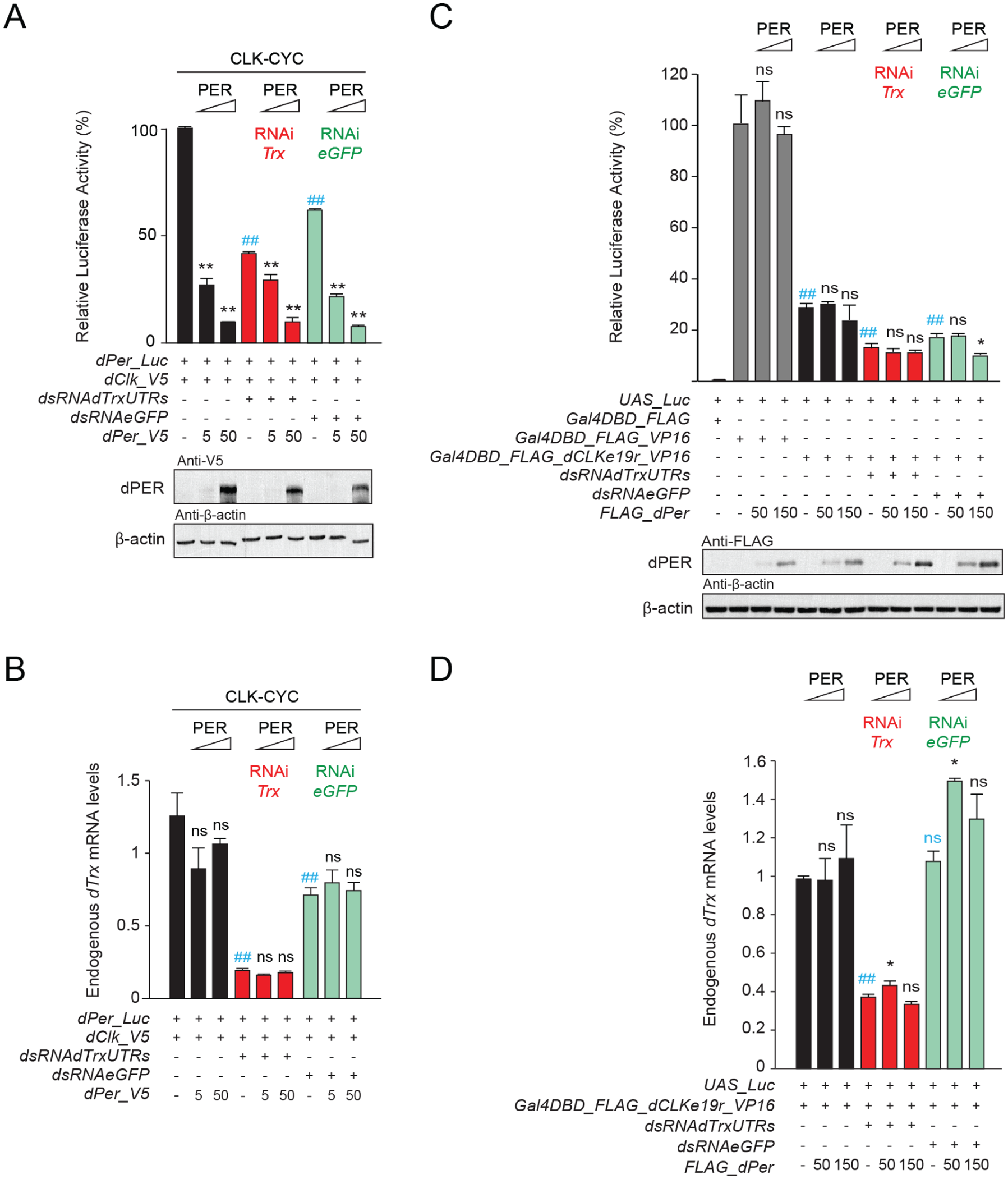
The histone methyltransferase TRX is necessary for CLK:CYCLE-mediated transcriptional activation but not PER repression. (a) The Drosophila per promoter-driven luciferase reporter (dPer_Luc; 10 ng) was expressed in Drosophila S2 cells with dCLK (5 ng) and increasing doses of dPER (amount given in ng) and in the presence (+) or absence (−) of double stranded (ds) RNA against egfp or the 5’ and 3’ UTRs of dTrx (7.5 µg each). Firefly luciferase activity was computed relative to renilla luciferase (30 ng) activity. Each value is mean ± SEM of 3 replicates. One-way ANOVA, Tukey post hoc: ##p < 0.01 (in gray for activation); **p < 0.01 (in black for repression). Western blots of V5-tagged dPER and beta actin are shown. (b) Quantification of endogenous dTrx mRNA levels showing efficient knockdown of dTrx by RNAi. Values are mean ± SEM of 3 individual samples. One-way ANOVA, Tukey post hoc: ##p < 0.01 (in gray for activation); ns is nonsignificant (in black for repression). (c) A UAS luciferase reporter (UAS_Luc; 10 ng) was used in the presence (+) or absence (−) of plasmids expressing Gal4DBD_FLAG or dCLKe19r fused to a N-terminal Gal4DBD_FLAG and a C-terminal VP16 transactivation domain (Gal4DBD_FLAG_dCLKe19r_VP16) (5 ng each) and increasing doses of dPER (amount given in ng), in the presence or absence of double-stranded (ds) RNA against egfp or the 5’ and 3’ UTRs of dTrx (7.5 µg each). Quantification of luciferase activity and statistics were performed as in (a). One-way ANOVA, Tukey post hoc: ##p < 0.01 (in gray for activation); *p < 0.05, and ns is nonsignificant (in black for repression). Western blots of FLAG-tagged dPER and beta actin are shown. (d) Quantification of endogenous dTrx mRNA levels showing efficient knockdown of dTrx by RNAi. Values are mean ± SEM of 3 individual samples. One-way ANOVA, Tukey post hoc: ##p < 0.01, ns is nonsignificant (in gray for activation); *p < 0.05, ns is nonsignificant (in black for repression). The ## symbol indicates the P-value for activation.
Previous work using Drosophila clock components showed that dCLKe19r is required for dPER binding via other regions of dCLK in S2 cells (Lee et al., 2016). Since TRX is required for dCLK activation but has minimal impact on dPER repression (Figures 1 and 2), we tested the extent to which TRX impacts dPER binding to dCLKe19r. We performed immunoprecipitations (IPs) with epitope tagged versions of dCLK, dCLKΔ19r and dPER expressed in S2 cells in which Trx was or was not knocked down via RNAi. Surprisingly, our results show that in the absence of dCLKe19r or the presence of Trx RNAi, PER-CLK binding is reduced but not eliminated (Figure 3). Since dPER can repress transcription in the absence of dCLKe19r or TRX (Figures 1 and 2), this result suggests that in contrast to mammals and monarchs, dPER binds other regions of dCLK to effect transcriptional repression in flies, consistent with previous results (Lee et al., 2016).
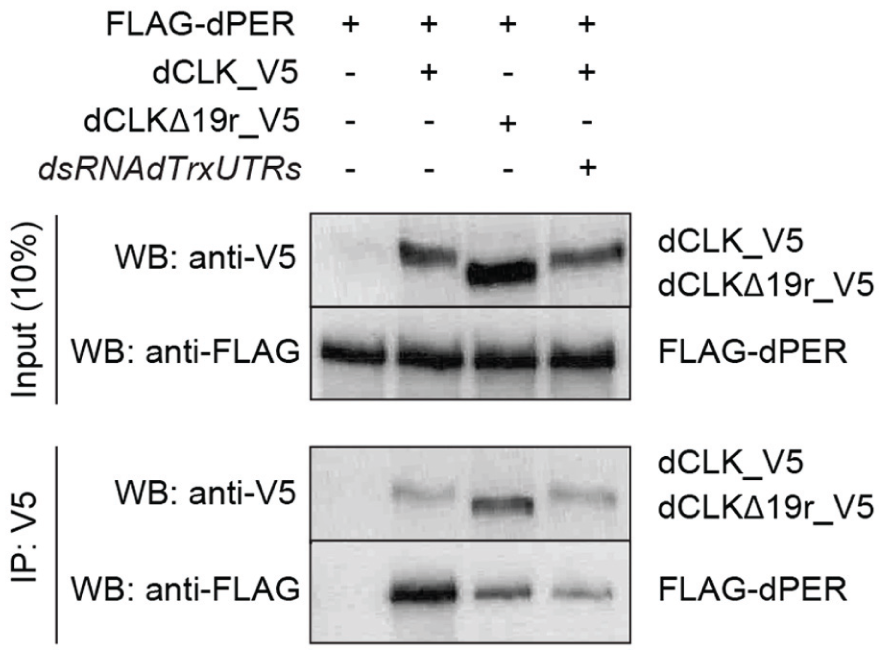
CLKe19r and the histone methyltransferase TRX contribute to CLK-PER interaction. Co-immunoprecipitations (IPs) from S2 cells transfected with FLAG_dPER and either dCLK_V5 or dCLKΔ19r_V5 expressing plasmids (400 ng each) in the presence (+) or absence (−) of dsRNA against dTrx 5’- and 3’-UTRs (7.5 µg each). Anti-V5 antibody was used for IPs and Western blots (WB) were probed with the indicated antibodies.
HSP68 Is Required for Repression by dPER
In monarchs, HSP68 is required for both dpCLK activation and dpPER repression (Zhang et al., 2022). However, TRX methyltransferase activity is required to directly or indirectly methylate HSP68 on R45 to promote dpPER repression but is not required for dpCLK activation (Zhang et al., 2022). Given the role HSP68 plays on transcriptional activation and repression in monarchs and that dPER repression persists in flies when Trx expression is greatly reduced (Figure 2), we reasoned that HSP68 would contribute to dCLK activation but not dPER repression in flies.
To determine whether this was the case we carried out transcription assays in S2 cells. dCLK-dependent activation of dPer-Luc was reduced by ~90% upon Hsp68 RNAi knock down but could be rescued to 40% to 50% of no Hsp68 RNAi controls by expressing either wild-type Hsp68 or an Hsp68 mutant that could not be methylated at R45 (i.e. Hsp68_R45A) (Figure 4a), demonstrating that HSP68 is necessary for dCLK activation independent of R45 methylation. However, dPER expression did not further decrease the already low levels of dPer-Luc expressed after Hsp68 RNAi knockdown but rather led to an anomalous increase in dPer-Luc levels when dPer was expressed (Figure 4a). Given these results, we tested whether boosting dPer-Luc expression using dCLK_VP16 would provide sufficient activation to determine whether knocking down Hsp68 expression alters dPER repression. Indeed, dCLK_VP16 increased dPer-Luc levels, and when Hsp68 expression was knocked down, dPER repression showed a small but significant decrease only after adding the highest amount of dPer-V5 (Figure 4b). The partial rescue of dCLK activation by wild-type Hsp68 and Hsp68_R45A allowed us to test whether HSP68 R45 methylation was required for dPER repression. Both wild-type HSP68 and HSP68_R45A rescued dPER repression to a similar extent as that seen in the absence of Hsp68 RNAi knockdown (Figure 4a), demonstrating that TRX-dependent methylation of HSP68 R45 is not required for dPER repression.
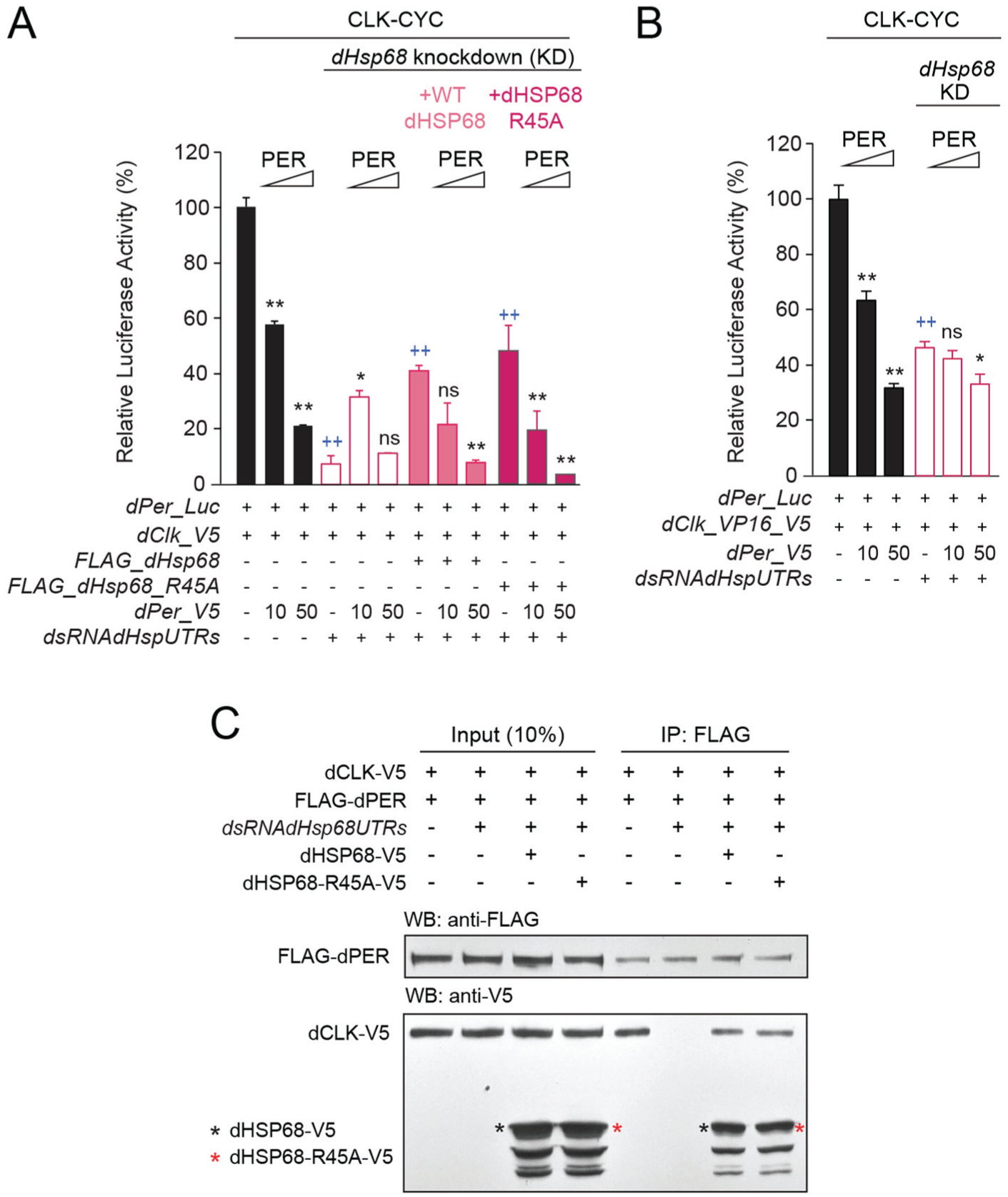
HSP68, but not its methylation on arginine 45, is necessary for CLK-CYCLE activation, PER repression, and CLK-PER interaction. (a) The Drosophila per promoter-driven luciferase reporter (dPer_Luc; 10 ng) was expressed in the presence of dCLK expression plasmids (5 ng each), increasing doses of dPER (amount given in ng), without or with dsRNA targeting endogenous dHsp68 5’- and 3’-UTRs (7.5 µg each), and plasmids expressing either wild-type or R45A mutant dHSP68 (50 ng each). Firefly luciferase activity was computed relative to renilla luciferase activity. Each value is mean ± SEM of 3 replicates. One-way ANOVA, Tukey post hoc: ++p < 0.01 (in gray for activation); **p < 0.01, *p < 0.05, and ns is nonsignificant (in black for repression). (b) dPer_Luc (10 ng) was used in the presence of dCLK_VP16_V5 to boost activation levels and increasing doses of dPER (amount given in ng), in the presence or absence of dsRNA targeting endogenous dHsp68 5’- and 3’-UTRs (7.5 µg each). Firefly luciferase activity was computed relative to renilla luciferase activity. Each value is mean ± SEM of 3 replicates. One-way ANOVA, Tukey post hoc: ++p < 0.01 (in gray for activation); **p < 0.01, *p < 0.05, and ns is nonsignificant (in black for repression). (c) Co-IPs from S2 cells transfected with dCLK_V5 and FLAG_dPER expressing plasmids (400 ng each), without or with dsRNA targeting dHsp68 5’- and 3’-UTRs (15 µg each), and with either wild-type or mutant R45A V5-tagged dHSP68 (400 ng each). Anti-FLAG antibody was used for IPs and Western blots (WB) were probed with the indicated antibodies. The ++ symbol indicates the P-value for activation.
HSP68 R45 methylation is required for dpCLK-PER binding in monarchs (Zhang et al., 2022), which likely explains why TRX is required for dpPER repression. Since HSP68 methylation is not required for dPER repression, we wanted to test whether wild-type HSP68 and/or HSP68-R45A were required for dCLK-PER binding. dPER was immunoprecipitated (IPed) from S2 cells expressing V5-tagged dCLK (dCLK-V5) and FLAG-tagged dPER (FLAG-dPER) alone, S2 cells expressing dCLK-V5, FLAG-dPER and Hsp68 RNAi, or S2 cells expressing dCLK-V5, FLAG-dPER, Hsp68 RNAi, and either wild-type V5-tagged HSP68 (HSP68V5) or V5-tagged HSP68-R45A (HSP68-R45A-V5). Inputs and IPs were then run on Western blots that were probed with FLAG and V5 antibodies. These results show that upon Hsp68 RNAi knockdown, dPER-CLK binding is eliminated (Figure 4c), indicating that HSP68 is required for dPER repression. The loss of dPER-CLK binding is rescued by either HSP68 or HSP68_R45A (Figure 4c), demonstrating that while HSP68 is necessary for dPER-CLK binding, its R45 methylation is not required. In addition, both HSP68 and HSP68_R45A associate with the dPER-CLK complex (Figure 4b), showing that HSP68 binds to dCLK, dPER, or both. Together, these results demonstrate that HSP68 plays a conserved role in mediating dCLK activation and dPER repression in both monarchs and flies, but TRX-dependent methylation of HSP68 R45 is only required for dpPER repression.
Discussion
CLK-dependent activation and PER-dependent repression drive the TFLs that keep circadian time in animals. Experiments in mammals, monarchs, and Drosophila identified the region of CLOCK/CLK encoded by exon 19 (e.g. CLKe19r) as a key domain involved in activating transcription (Katada and Sassone-Corsi, 2010; Lee et al., 2016; Zhang et al., 2022). Studies in mammals and monarchs showed that the CLOCK/CLKe19r is bound by MLL1/TRX, which methylates histone H3 to activate CLOCK-BMAL1/CLK-BMAL1 transcription. Here, we confirm previous work demonstrating that dCLKe19r is required for dCLK activation (Figure 1a) and further show that TRX also contributes to dCLK activation in flies (Figure 2a and 2b). Thus, this and previous studies collectively show that TRX plays a conserved role in promoting CLK activation via CLKe19r binding.
We then assessed whether the dCLKe19r and TRX also contribute to dPER repression. In contrast to monarchs, the dCLKΔ19r mutant which lacks the dCLKe19r has little impact on dPER transcriptional repression in S2 cells (Figure 1a and 1b). This result implies that dPER binds other portions of dCLK to effect repression, consistent with previous work showing that dPER interacts with at least one other portion of dCLK (Lee et al., 2016). TRX binds the dpCLKe19r to effect dpPER repression in S2 cells (Zhang et al., 2022), thus given that expressing the dCLKΔ19r mutant in S2 cells had little impact on dPER repression we reasoned that RNAi knockdown of Trx would also not greatly alter dPER repression. Indeed, dPER repression persists when Trx expression is knocked down (Figure 2a). Consistent with this result, dPER is unable to repress via the dCLKe19r either in the presence or absence of Trx RNAi knockdown (Figure 2b), confirming that TRX does not contribute to dPER repression of dCLK transcription. Interestingly, dPer-Luc levels in cells expressing Gal4DBD_FLAG_dCLKe19r_VP16 are ~70% lower than in control cells expressing Gal4DBD_FLAG_VP16 (Figure 2b). Since the dCLK repressor CIPC also binds the dCLKe19r (Hou et al., 2017), the lower levels of dPer-Luc expression in cells expressing Gal4DBD_FLAG_dCLKe19r_VP16 may be due to CIPC binding. Although the dCLKe19r and TRX are not necessary for dPER repression, both promote dPER-CLK binding; the dCLKΔ19r mutant and Trx RNAi knockdown greatly reduce, but do not eliminate, dPER-CLK binding (Figure 3). Thus, even though the dCLKe19r and TRX both contribute to dPER-CLK binding, dPER presumably binds other portions of dCLK to effect transcriptional repression.
Our previous work in monarchs showed that HSP68 is required for dpCLK activation and for dpPER repression (Zhang et al., 2022). We initially tested whether HSP68 contributes to dCLK activation by measuring dPer-Luc levels in the presence or absence of Hsp68 RNAi in S2 cells. We found that RNAi knockdown of Hsp68 reduced dCLK activation by ~90% and dCLK_VP16 activation by ~2-fold (Figure 4). Although the Hsp68 RNAi we used targeted 444 bp of the 5’-UTR and 416 bp of the 3’-UTR, it was also capable of binding other members of the HSP70 gene family, albeit less effectively as RNAi binding covered smaller or less homologous regions. Thus, reduced levels of dCLK activation may result from lower expression levels of Hsp68 and likely other Hsp70 family members, indicating that HSP68 and possibly other HSP70s are necessary for robust dCLK transcription. However, expressing wild-type Hsp68 partially rescues dCLK activation by more than 4-fold, demonstrating a role for HSP68 in dCLK activation (Figure 4). It is possible that HSP68 is unable to fully rescue dCLK activation because expression of other HSP70s is reduced. dCLK activation was also partially rescued by expressing the Hsp68 R45A mutant (Figure 4), demonstrating that HSP68 methylation is not required for activation. These experiments show that HSP68 plays a conserved role in promoting dCLK activation.
We show that HSP68 is also required for dPER repression. Although dPER did not reduce the very low levels of dPER-Luc further after Hsp68 RNAi knockdown, when we used dCLK_VP16 to boost dPer-Luc expression, knocking down Hsp68 expression greatly reduced dPER repression of CLK-CYC activation (Figure 4b). However, dCLK repression by dPER was rescued by expressing wild-type HSP68 or the HSP68 R45A mutant in Hsp68 RNAi knockdown cells, demonstrating that HSP68 expression is necessary for dCLK repression independent of HSP68 methylation (Figure 4a). In addition, Hsp68 RNAi knockdown eliminates dPER-CLK binding in S2 cells, whereas expressing either wild-type HSP68 or the HSP68 R45A mutant in Hsp68 RNAi knockdown cells restores dPER-CLK binding (Figure 4c). Both wild-type HSP68 and the HSP68 R45A mutant are present in the dPER IP (Figure 4c), indicating that HSP68 is a component in this repression complex. The ability of the HSP68 R45A mutant to rescue dPER repression in Hsp68 RNAi knockdown cells contrasts with the situation in monarchs, where HSP68 R45 must be methylated to effect dpPER repression (Zhang et al., 2022). Given that HSP68 R45 methylation is dependent on TRX in monarchs, it is not surprising that HSP68 R45 methylation is not necessary for dPER repression as TRX is not required for dPER repression (Figure 2). These experiments demonstrate that HSP68 (and perhaps other HSP70s) are required for dPER repression by facilitating dPER-CLK binding as a component of the dPER repression complex.
Taken together with previous results in mammals and monarchs, our results using Drosophila clock components show that the CLKe19r and TRX play a conserved role in promoting dCLK activation (Figure 5), presumably through H3K4 trimethylation (Katada and Sassone-Corsi, 2010). Likewise, HSP68 plays a conserved role in promoting CLK-BMAL1 transcription in monarchs (Zhang et al., 2022) and CLK-CYC transcription in Drosophila (Figures 4 and 5), though the role HSP68 plays has not been identified. Given that HSP68 is an ATPase that functions to fold proteins (Hartl and Hayer-Hartl, 2009; Kampinga and Craig, 2010), we suspect that HSP68 is involved in folding of proteins required for dPER-CLK binding. In contrast to monarchs, dCLKe19r and TRX are not required for dPER repression (Lee et al., 2016) (Figure 2), demonstrating that dCLK transcriptional repression operates to some extent via other regions of dCLK that interact with dPER (Lee et al., 2016) (Figure 3). TRX effects repression in monarchs by directly or indirectly methylating HSP68 R45 (Zhang et al., 2022), but such TRX-dependent HSP68 methylation is not required for dPER repression in Drosophila (Figures 4 and 5). Based on previous data and that described here, the CLKe19r, MLL1/TRX, and HSP68 play conserved roles in the animal circadian clock by promoting CLOCK/CLK transcription (Figures 1-4) (Katada and Sassone-Corsi, 2010; Lee et al., 2016; Zhang et al., 2022). However, Drosophila differs from monarchs in that TRX is not required for dPER repression yet marginally contributes to dPER-CLK binding (Figures 2a, 2c, and 3). The lack of TRX involvement in dPER repression may be related to the fact that HSP68 R45 methylation is unnecessary in flies but is critical in monarchs. This connection between TRX and HSP68 function in monarchs reveals a mechanistic difference in PER repression, but additional experiments are needed to determine whether this represents a broader difference between PER-TIM repression and PER-CRY2 repression. Despite the difference in HSP68 methylation between monarchs and Drosophila, it is clear that HSP68 (and perhaps other HSP70 paralogs) play a conserved role in PER repression just as they do in CLK activation (Figure 5). Future work will investigate the role HSP68 plays to control TFL function in animals.
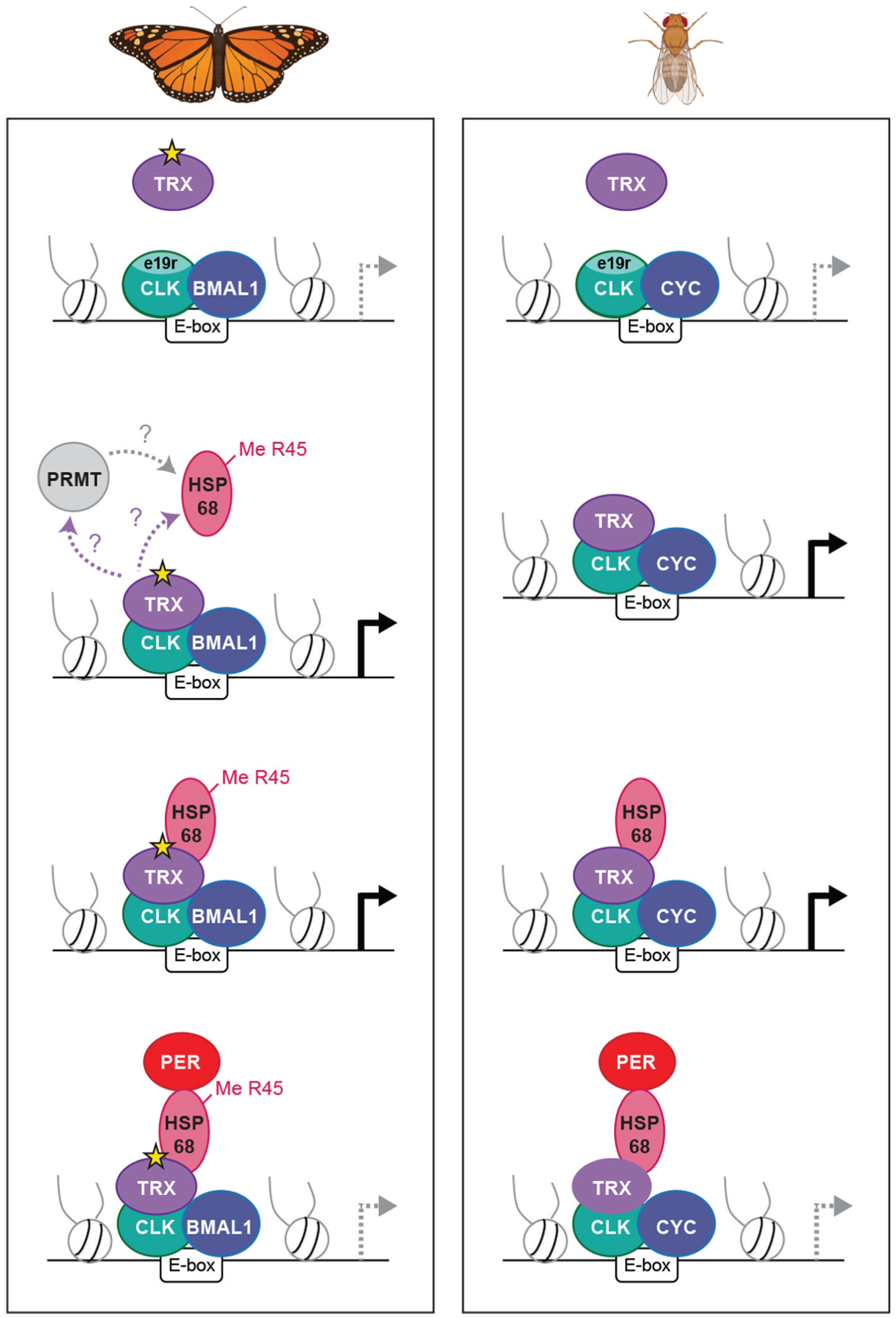
Model comparing the roles of TRX and HSP68 for CLK activation and PER repression in the monarch butterfly and Drosophila clocks. The CLK exon 19 region (e19r), TRX, and HSP68 are all required for monarch CLK-BMAL1 and Drosophila CLK-CYC transcriptional activation. In the monarch, TRX and HSP68 are also required for PER repression whereby TRX catalytically (gray star) methylates HPS68 on arginine 45 (R45) either directly or indirectly via the recruitment of a protein arginine methyltransferase (PRMT); modified from Zhang et al. (2022). In Drosophila, the PER repression mechanism retains HSP68 function but does not require TRX-dependent methylation.
Supplemental Material
sj-xls-1-jbr-10.1177_07487304251386708 – Supplemental material for TRITHORAX and HSP68 Regulate Clock Gene Transcription in the Drosophila Transcriptional Feedback Loop
Supplemental material, sj-xls-1-jbr-10.1177_07487304251386708 for TRITHORAX and HSP68 Regulate Clock Gene Transcription in the Drosophila Transcriptional Feedback Loop by Ying Zhang, Paul E. Hardin and Christine Merlin in Journal of Biological Rhythms
Footnotes
Acknowledgements
We thank Michael Rosbash (Brandeis University) for sharing the pAC-dPer_V5, dPer_Luc, and pACT-Renilla-Luc plasmids, Matthew Sachs (Texas A&M University) for the use of the VICTOR3V Multilabel Plate Counter, Jerome Menet (Texas A&M University) for discussions, and Nirav Thakkar and Minal Jain (Texas A&M University) for feedback on the manuscript. This work was supported by NIH grant R01 GM124617 (to C.M. and P.E.H.).
Conflict of Interest Statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Christine Merlin and Paul E. Hardin are members of the Editorial Board of Journal of Biological Rhythms. The authors did not take part in the peer review or decision-making process for this submission and has no further conflicts to declare.
Materials Availability
All unique reagents generated in this study are available from the corresponding authors without restriction.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.