
Abstract

“Ten percent of the expressed genes are rhythmic” has become an oft-repeated yardstick of circadian transcriptomics of any given tissue. Moreover, a large fraction of expressed genes were rhythmic in only one tissue, leading to the understanding that circadian regulation is highly tissue-specific (Zhang et al., 2014). The focus on improving rhythm detection to eke out every rhythmic gene has led to newer methods capable of finding rhythms with more general waveforms, while making fewer assumptions (Hughes et al., 2017). This appropriate but disproportionate emphasis on statistical significance has been realized at the cost of clarity on the biological significance of the identified rhythms—an all-too-common challenge in computational life sciences. For example, have we paused to wonder whether this “consensus” circadian transcriptome is robust in number and is consistent in identity across different studies and labs?
Brooks et al. (2023) in this issue did just that. They performed a meta-analysis of liver transcriptomic (RNA-seq) data sets of normal mice collected under standard conditions (ad-lib or night-restricted feeding and constant dark or LD conditions). Rather disappointingly, variation from technical factors (study design, sequencing technology) exceeded the biological differences (sex, time) between samples and studies. The circadian rhythms of core clock genes were remarkably consistent in phase and, to a large extent, amplitude as has been previously observed. Brooks et al. chose a fixed rhythm detection algorithm (JTK Cycle) and focused on comparing the results of rhythmicity analysis across the 57 studies included in the meta-analysis.
The overlaps in lists of rhythmic genes across studies were marginal with the largest overlaps between studies with the highest number of time samples. This is expected as (a) we have shown previously that comparing lists of rhythmic genes (across studies) falsely overestimates the degree of divergence of the lists (Pelikan et al., 2022) and (b) the number of false positives decreases (and power increases) with increasing number of time samples (per cycle and across cycles). Direct pairwise comparison of rhythm patterns across studies found only a few statistically significant differences, suggesting that although the lists differed greatly, the data itself do not differ significantly. None of these qualitative conclusions changed when a better rhythm detection algorithm (BooteJTK) was used instead. Moreover, the phase distributions of these widely divergent rhythmic gene sets were also inconsistent. Finally, using the consistency in the raw data across studies, the authors performed shape-invariant rhythm analysis combining the 57 studies to find 2712 rhythmic shapes including core clock genes and several complex waveforms (asymmetric or with multiple peaks) that are typically inaccessible with the limited time sampling in individual studies.
So where does that leave us in our quest to quantify the circadian transcriptome? Except for studies designed for high statistical power, the list of rhythmic genes is not reproducible, and hence unreliable, irrespective of the rhythm detection approach. Even so, comparing rhythms across conditions rather than cataloging rhythmic genes in one condition in isolation is likely to yield more robust and consistent results (Figure 1). Brooks et al. raise further questions about studying the transcriptional output of the clock. Ought we to do away with quantifying rhythmicity of genes and directly evaluate rhythmicity of pathways or functional modules (e.g., Winkler et al., 2022)? If there is such poor consistency across circadian transcriptomes of the same tissue (liver), what does that mean for our understanding of tissue-specificity of rhythms? Or, for the extent of actual discordance between transcriptional and protein rhythms? A theory on the origin of these genome-wide rhythms is also essential to design algorithms to detect biologically relevant rhythms. I am biased towards the theory that “everything is rhythmic” in the transcriptome and natural selection coordinated only the necessary “circadian” pathways to be coherent, while irrelevant pathways just have uncoordinated low-amplitude rhythms. In this vein, it is possible that a lot of the detected inconsistent rhythms are simply context-specific and biologically irrelevant. An alternate theory suggests that selection imbued pathways (and genes) with high-amplitude rhythms leaving the rest arrhythmic. So, coming full circle, the 10% rule of thumb is merely that—a consensus that arose from many early transcriptomic studies that just happen to have similar time sampling designs—and a list of rhythmic genes provides only limited functional insight into the output of the clock.
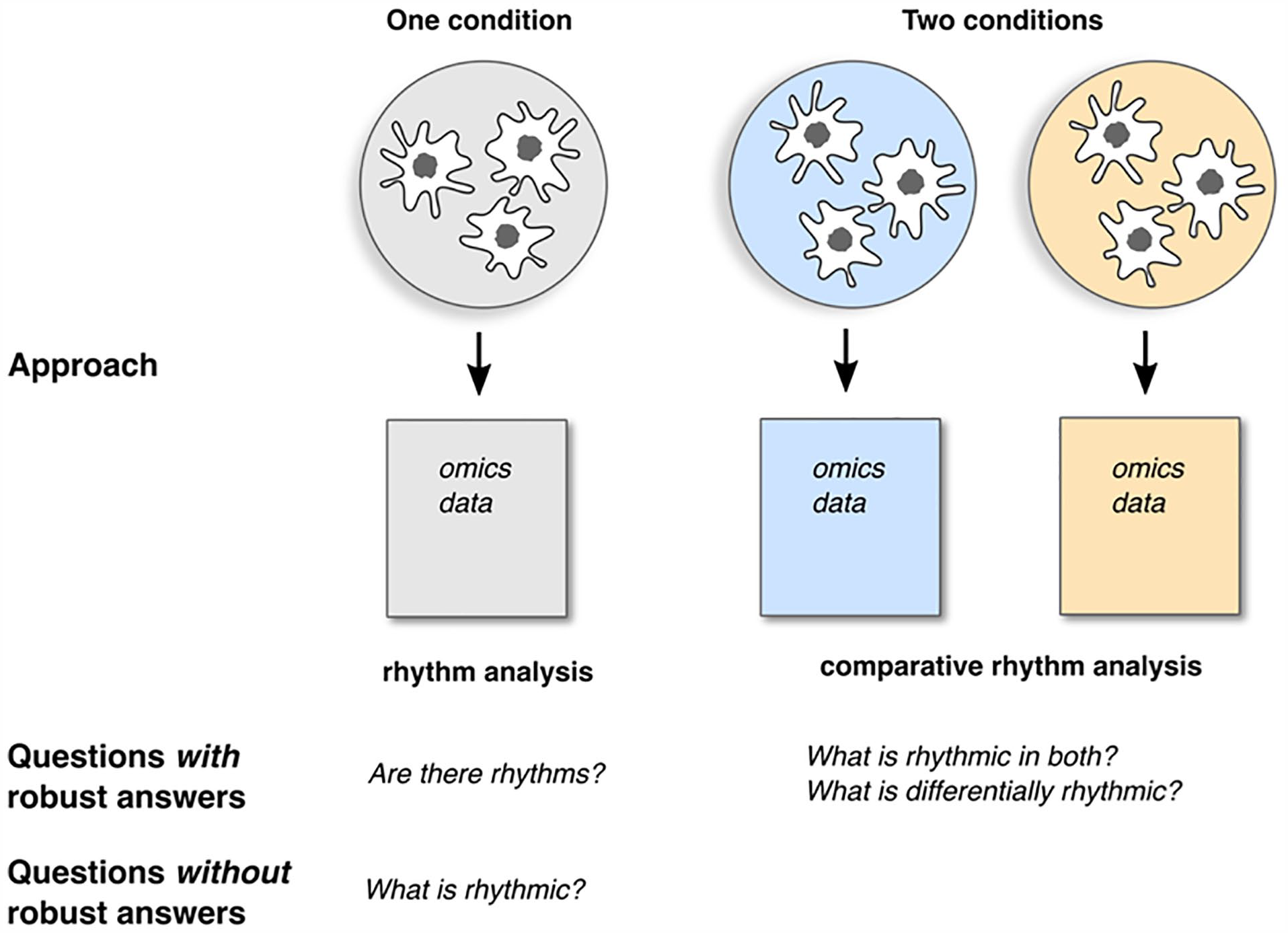
Limitations of experimental designs to quantify circadian rhythms in high-throughput data.