
Abstract
The endogenous circadian period of animals and humans is typically very close to 24 h. Individuals with much longer circadian periods have been observed, however, and in the case of humans, these deviations have health implications. Previously, we observed a line of Drosophila with a very long average period of 31.3 h for locomotor activity behavior. Preliminary mapping indicated that the long period did not map to known canonical clock genes but instead mapped to multiple chromosomes. Using RNA-Seq, we surveyed the whole transcriptome of fly heads from this line across time and compared it with a wild-type control. A three-way generalized linear model revealed that approximately two-thirds of the genes were expressed differentially among the two genotypes, while only one quarter of the genes varied across time. Using these results, we applied algorithms to search for genes that oscillated over 24 h, identifying genes not previously known to cycle. We identified 166 differentially expressed genes that overlapped with a previous Genome-wide Association Study (GWAS) of circadian behavior, strongly implicating them in the long-period phenotype. We tested mutations in 45 of these genes for their effect on the circadian period. Mutations in Alk, alph, CG10089, CG42540, CG6034, Kairos (CG6123), CG8768, klg, Lar, sick, and tinc had significant effects on the circadian period, with seven of these mutations increasing the circadian period of locomotor activity behavior. Genetic rescue of mutant Kairos restored the circadian period to wild-type levels, suggesting it has a critical role in determining period length in constant darkness.
Circadian rhythms are ubiquitous, exhibited at the molecular, biochemical, physiological, behavioral, and population levels across all organisms (Merrow et al., 2005; Huang et al., 2011). These biological processes typically fluctuate over 24 h, and are under partial genetic control (Emery et al., 1995; Hofstetter et al., 1995; Harbison et al., 2019; Keenan et al., 2020). Both naturally occurring and chemically induced EMS (ethyl methanesulphonate) mutations in canonical clock genes can produce free-running locomotor period lengths significantly deviant from the characteristic 24-h circadian rhythm (Konopka and Benzer, 1971; Rothenfluh et al., 2000; Tauber et al., 2007). For instance, a missense point mutation in the Per-Arnt-Sim domain encoding region of the canonical clock gene period (per) lengthened the circadian period to 28.6 h in constant darkness (Konopka and Benzer, 1971; Hardin et al., 1990). Period lengthening mutants of timeless (tim) have also been identified using forward genetic screens, with periods ranging from 26 to 28 h (Rothenfluh et al., 2000). Thus, mutations in a single canonical clock gene can substantially increase the circadian period.
In Drosophila melanogaster, circadian rhythms require oscillations in the transcripts of canonical clock genes; accordingly, the transcripts of per and tim oscillate over a 24-h cycle (Merrow et al., 2005; Huang et al., 2011). A previous study showed an inverse relationship between per transcript levels and the behavioral periodicity—that is, flies with low levels of per transcript levels displayed longer periods and those with higher per abundance exhibited shorter periodicities in their locomotor activity (Baylies et al., 1987). Thus, transcript levels of canonical genes play an important role in determining/influencing the length of circadian rhythms in Drosophila.
We recently observed flies with a very long circadian period in the process of conducting a Genome-wide Association Study (GWAS) of circadian behavior in the Drosophila Genetic Reference Panel (DGRP; Harbison et al., 2019). DGRP_892 is a wild-derived inbred line of the DGRP with an average circadian period of 31.3 h (Harbison et al., 2019). Unlike the known canonical clock polymorphisms producing a long period, preliminary mapping suggested that the long-period phenotype mapped to multiple regions in the genome (Harbison et al., 2019). Importantly, DGRP_892 was removed from the genome-wide association analysis as the long-period phenotype was an outlier. The remaining lines of the DGRP were used to compute the genotype-phenotype associations. The GWAS identified 584 SNPs tagging 268 genes associated with circadian period and rhythmicity index (Harbison et al., 2019).
In principle, the genes contributing to the long-period phenotype in DGRP_892 could be among the 268 identified in the GWAS. However, picking the most promising genes from a large list of potential candidates remains one of the challenges of functional genomics. One way to prioritize candidate genes for further study is to choose those genes repeatedly identified among disparate data analyses. This systems genetics approach has been applied previously to identify gene networks affecting sleep, sleep deprivation, and recovery sleep in mice, where the results of quantitative trait locus (QTL) mapping, expression QTL mapping, predicted annotation effects, differential gene expression analyses, and quantitative trait transcript mapping were combined to prioritize genes (Diessler et al., 2018). Transcriptional expression QTL mapping data were also used to refine large genomic QTL for sleep in flies (Smith and Macdonald, 2020). Thus, combinatorial systems genetics approaches can assist in the prioritization of important candidate genes for complex traits.
With this approach in mind, we surveyed the transcriptome of whole heads in DGRP_892 over time and compared it with the transcriptome of a line with a 24-h circadian period. We identified transcripts differentially expressed among genotype, time, sex, and their interactions. We then compared the transcriptional data with the previous GWAS (Harbison et al., 2019). We found that 62% of the genes identified by the GWAS also had genes differentially expressed between the two genotypes. In addition, using RAIN and JTK_CYCLE, we identified transcripts with putatively aberrant cycling in DGRP_892. These combined analyses suggested 166 candidate genes that might affect circadian period.
Of the 166 candidate genes, 45 had Minos insertion lines available for testing. We assessed the circadian period of these Minos insertions and confirmed 11 genes with significant differences in circadian period as compared with the isogenic control. These genes may therefore contribute to the long period in DGRP_892. In addition, we rescued one of the long-period phenotypes by precise excision of a Minos element in CG6123, a gene we have renamed Kairos. In this way, we demonstrate that the relatively common variants we identified in the DGRP have altered regulation of gene expression in DGRP_892 that may explain its long circadian period.
Materials and Methods
Drosophila Stocks
We investigated gene expression in DGRP_892, a line from the DGRP (Mackay et al., 2012). We previously demonstrated that DGRP_892 has a very long circadian period; on average, the circadian period was 31.3 h in these flies (Harbison et al., 2019). We used w1118; Canton-S B (CSB) as a control line; these flies have an average circadian period of 23.9 h (Harbison et al., 2019).
To confirm the genes that we identified, we tested circadian behavior in Minos element mutants targeting 45 candidate genes. These transposable element mutations are readily available from the Bloomington, IN, stock center (BDSC, Indiana University) and have isogenic control lines (Bellen et al., 2011). Ten Minos elements targeted genes with expression that was predicted to cycle and overlapped previous GWAS results (Harbison et al., 2019). We tested an additional 35 candidate genes that were differentially expressed between the DGRP_892 and CSB genotypes and overlapped the previous GWAS results (Harbison et al., 2019). Supplementary Table S1 lists the mutant lines tested.
Sample Collection for RNA-Seq
Flies were maintained under standard culture (cornmeal-sucrose-agar medium, 25 °C, 60%-75% relative humidity) and lighting conditions (12-h light: dark cycle; LD). Five virgin males and five females were used to set up w1118, CSB, and DGRP_892 cultures in an LD incubator. Soon after eclosion, 24 separate-sex vials with 150 virgin males and 150 virgin females per vial were collected for each genotype. One set of flies was entrained in an incubator with a 12:12 LD cycle (0600 h “lights-on”; 1800 h “lights-off”) and another set was entrained in an incubator with a reverse LD cycle (1800 h “lights-on” and 0600 h “lights-off”) for 3 days. Both groups of flies were then transferred to a common incubator in constant darkness (DD). On the third day of DD, flies were flash-frozen on dry ice every 2 h starting at circadian time (CT02/CT12) and maintained at ‒80 °C until further processing. Flies were decapitated on dry ice for RNA extraction. We replicated this procedure three times to produce 144 RNA samples (three biological replicates, two sexes, two genotypes, and 12 time points).
RNA Extraction
Samples were randomized and extracted in groups of 12. For each RNA sample, 150 heads were placed in Omni-ruptor tubes (Omni International, Kennesaw, GA) with four RNase/DNase-free metal beads (Omni International, Kennesaw, GA) and homogenized in 1 mL TRIzol (Thermo Fisher Scientific, Grand Island, NY) using an Omni-bead ruptor with the following settings: Speed, 5.65 m/s; Time, 20 s; Duration, 30 s; Cycles, 2. Homogenates were incubated at room temperature for 5 min; 200 µL of chloroform (Mallinckrodt Baker, Center Valley, PA) was added to each tube; samples were then mixed by shaking for 15 s, incubated at room temperature for 3 min, and centrifuged at 4 °C, 12,000 × g for 15 min. The 500-µL aqueous phase was transferred to new tubes and RNA was further purified using the PureLink RNA Mini Kit (Life Technologies, Carlsbad, CA) with on-column DNase treatment, according to the manufacturer’s instructions. Purified total RNA was eluted in 60 µL of water and stored at ‒80 °C. RNA was quantified by NanoDrop 8000 (Thermo Fisher Scientific, Grand Island, NY) and quality verified by BioAnalyzer 2100 using the Agilent RNA 6000 Pico Kit (Agilent Technologies, Santa Clara, CA).
RNA Library Preparation and Sequencing
Prior to poly(A) RNA selection, 0.02 µL ERCC (External RNA Controls Consortium) Spike-in Control Mix 1 (Life Technologies, Carlsbad, CA) was added to 1 µg total RNA. Poly(A)-selected stranded mRNA libraries were constructed from 1 µg total RNA using Illumina TruSeq Stranded mRNA Sample Prep Kits (Illumina, San Diego, CA) per the manufacturer’s instructions with the following exception: Polymerase chain reaction (PCR) amplification was performed for 10 cycles rather than 15 to minimize the risk of over-amplification. Unique barcode adapters were applied to each library; dual indices were used. Libraries were pooled for sequencing.
The pooled libraries were sequenced on multiple lanes of a HiSeq4000 using version 4 chemistry to achieve a minimum of 12 million 76-bp read pairs. The sequences were processed using RTA version 1.18.64 and CASAVA 1.8.2 (Illumina, San Diego, CA).
Alignment
Quality Control (QC) of FASTQ files were assessed using FastQC toolkit (v0.11.5) using default parameters (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The paired-end reads were aligned against the D. melanogaster reference genome (dmel.r6.16; dos Santos et al., 2015) using STAR (v2.5.3a) alignment software with the following parameters “–twopassMode basic –sjdbOverhang 75” (Dobin et al., 2013). sjdbOverhang is the parameter for the length of the donor/acceptor sequence on each side of the junctions. Gene-level read counts were produced by featureCounts (v1.5.2; Liao et al., 2013, 2014) using only uniquely mapped reads with setting “-p -s 2” (paired-end, reverse-stranded). We used the trimmed mean of M-values (TMM) normalization method to normalize raw read counts (Robinson and Oshlack, 2010).
Sequence Analysis
We computed the distribution of TMM-normalized read counts mapping to either genic or intergenic regions. We used the 95th percentile of that distribution to detect the threshold for removing low count genes, which was 4.67 log2(TMM-normalized counts + 1) (Lin et al., 2016b). Genes with read counts smaller than the threshold for all samples were discarded from further analysis. Using the ERCC spike-in reads, we noted a bias in the data according to the date of the sequencing run and confirmed it in the sequence data using principal components analysis (Suppl. Fig. S1). We accounted for this bias with an additional parameter in our models (see below). We performed a likelihood ratio test using DESeq2 (nbinomLRT) to identify differentially expressed genes (Love et al., 2014). We made the following comparisons for each gene i.
To identify genes with significant differential gene expression across time points, we compared the full model
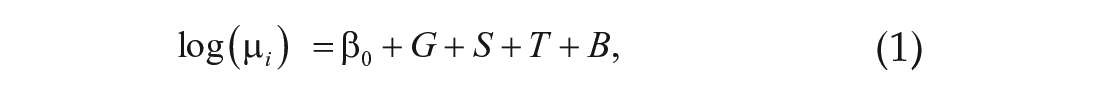
with the reduced model
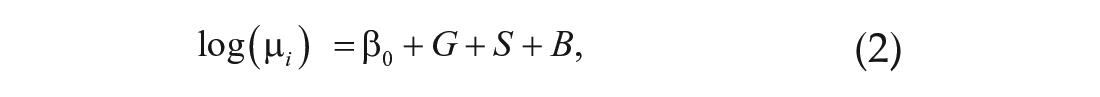
where G is genotype (DGRP_892 or CSB), S is sex, T is time, and B is batch effect due to sequencing date. We identified genes with significant differential expression across genotype and sex in the same manner. We identified significant two-way interactions among these terms (i.e., G × S, G × T, and S × T) by comparing the full model
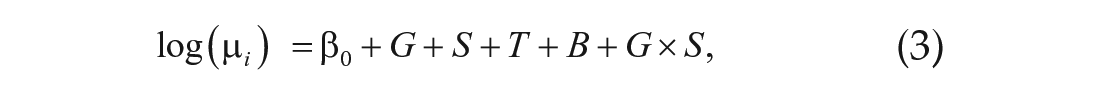
with the reduced model given by Equation 1;
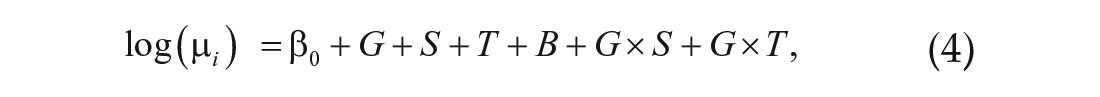
with the reduced model given by Equation 3; and
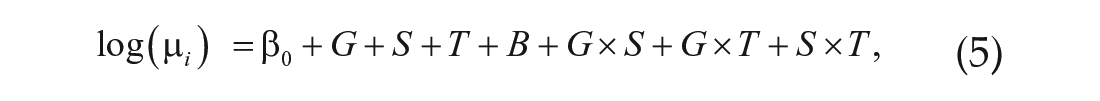
with the reduced model given by Equation 4. Finally, we identified significant three-way interactions (G × S × T) by comparing the full model
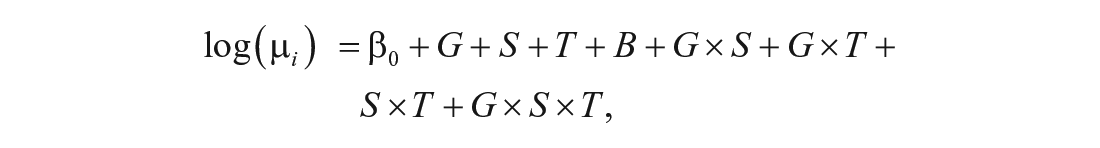
with the reduced model given by Equation 5. Adjusted p values were calculated using the software default, which uses the Benjamini-Hochberg method to determine false discovery rates (FDRs; Benjamini and Hochberg, 1995). Our threshold was an adjusted p value of 0.05 or less.
Genes with significant differential expression were further classified for potential gene-gene interactions using Modulated Modularity Clustering (MMC; Stone and Ayroles, 2009). MMC calculates the correlation of gene expression between any two gene pairs. The magnitude of the correlation is presumed to reflect the level of interaction among gene pairs. MMC applies an iterative graphical method to cluster correlated genes into highly interconnected groups. The MMC algorithm determines the number of highly interconnected clusters (or “modules”) rather than relying on a user-selected number of clusters. The correlations are re-ordered and graphed with the most highly interconnected groups of genes presented at the top left, while the least interconnected genes are graphed at the bottom right.
We further analyzed genes having significant differential expression among model terms containing time (i.e., T, G × T, and G × T × S) for possible circadian cycling using the JTK_Cycle and RAIN packages (Hughes et al., 2010; Thaben and Westermark, 2014). First, we removed the batch effect in our dataset from TMM-normalized reads using the ComBat function in the sva R package (Leek and Storey, 2007): (ComBat(data, batch = date, mod = “geno + sex + time”). Using RAIN, DGRP_892 and CSB were analyzed separately by sex with the following settings, rain(t(data), deltat = 2, period = 24, nr.series = 3, period.delta = 0, peak.border = c(0.35,0.65)). In JTK_Cycle, we used the following settings: (jtkdist(12,3) jtk.init[10:12,2])) for 12 samples, three replicates, min_period: 20, max_period: 24. These settings enabled us to search for rhythmic gene expression over a 24-h period. We repeated this analysis but adjusted the parameters in JTK_Cycle and RAIN to search for gene expression that was potentially cycling at longer periods, up to 32 h. The Benjamini-Hochberg procedure was used to control the FDR.
Creation of Minos Precise Excision Line
We followed the protocol for Minos element excision as previously reported to excise the insertion in CG6123 (Metaxakis et al., 2005). For the generation of excision events in the germ line of flies with single Minos insertions, flies homozygous for a Mi[ET1] transposon insertion on the X chromosome (Mi[ET1]CG6123[MB02356]) were crossed with flies carrying helper chromosome w[1118]; sna[Sco]/SM6a, P[w[+mC] = hsILMiT]2.4 (F1). Two days after setting up the crosses, the parent flies were removed and larvae in the vials were heat-shocked for 1 h per day for the next 3 days in a 37 °C water bath. Transformed progeny were screened for the presence of mosaic green fluorescent protein (GFP) eyes, indicating a partially transposed Minos element. Individual transformed males were backcrossed to females from the original Minos insertion line, CG6123[MB02356] (F2). The female progeny of this cross were heterozygous flies containing one copy of the GFP-positive Minos insertion and one copy of the GFP-negative putative excision chromosome, while GFP-negative males could be selected for directly. These progeny were crossed, and GFP-negative progeny were selected to create homozygous excision lines. DNA was extracted from the cultures to confirm precise excisions via PCR amplification and sequencing. We analyzed the Mi[ET1] excision event by PCR using a set of oligos flanking the original insertion site. The oligo sequences used were Forward Primer1-ttggttgcggagtaagtacg; Reverse Primer1-tgcgaatacagatgcggata, Forward Primer2 CATGGCCTTTGAACAGGATT; Reverse Primer2-TGCGAATACAGATGCGGATA, MI.3. Forward Primer-ATGATAGTAAATCACATTACG; MI.3.Reverse Primer-CAATAATTTAATAATTTCCC, MI.5.Forward Primer-CAAAAGCAACTAATGTAACGG; MI.5.Reverse Primer-TTGCTCTTCTTGAGATTAAGGTA.
Validation of Candidate Genes and Precise Excision Line
Flies were maintained under standard culture (cornmeal-sucrose-agar medium, 25 °C, 60%-75% relative humidity) and lighting conditions (12-h light: dark cycle (LD)). Five virgin males and five virgin females were used to set up cultures in an LD incubator. Virgin flies were collected and maintained in sex-separate vials for 3 days at LD. Individual flies were then transferred to DAM2 monitors (Trikinetics, Waltham, MA). Flies were placed into DAM monitor tubes with 5% sucrose, 2% agar food. Flies were monitored for 4 days in LD followed by 14 days in DD to assess their entrainment and free-running behavior, respectively. Sixteen flies per sex per line were tested. We calculated circadian period using the ClockLab program (Actimetrics, Wilmette, IL). We analyzed the data using the analysis of variance (ANOVA) model Y = µ + G + S + G × S + ε, where G is genotype and S is sex. For the precise excision line, we compared the circadian period of the excision line (CG6123MB*), the heterozygote (CG6123MB*/MB02356), the homozygous mutant line (CG6123MB02356), and the w1118 control with a post hoc Tukey test. We retested Minos and precise excision lines having significant genotypic effects, and analyzed the data with the ANOVA model Y = µ + G + S + R + G × S + G × R + R × S + G × S × R + ε, where G and S are as defined above and R is replicate.
Data Availability
All RNA-Seq data from this study are available from the National Center for Biotechnology Information (NCBI) GEO under the accession number GSE155018, including raw read counts and batch-adjusted TMM-normalized read counts for each gene per genotype/replicate/sex. All supplemental figures and tables are available online along with the article.
Results
Previously, we identified a line of the DGRP with an unusually long circadian period of 31.3 h (Harbison et al., 2019). We collected and sequenced RNA from fly heads of this line, DGRP_892, and compared it with a line with a 24-h circadian period, CSB. We collected the RNA at 2-h intervals over a 24-h period to identify cycling transcripts. Figure 1 shows the experimental setup: three replicate biological samples were collected according to genotype, time point, and sex. We found 11,474 transcripts with detectable expression in fly heads. Using a generalized linear model, we identified genes that were differentially expressed by Genotype, Time, and Sex, and their interactions. Table 1 lists the numbers and percentages of differentially expressed genes according to the experimental factor. Normalized read counts for each gene and additional details can be found in Supplemental Table S2. In total, we found 10,119 genes with differential expression (FDR adjusted p value < 0.05). Increasing the stringency of the FDR adjusted p value to 0.01 or 0.001 (Suppl. Table S3) had little effect on the numbers of differentially expressed genes among Genotype or Sex but did affect model terms containing the environmental factor of Time, as might be anticipated from previous work (Lin et al., 2016b).
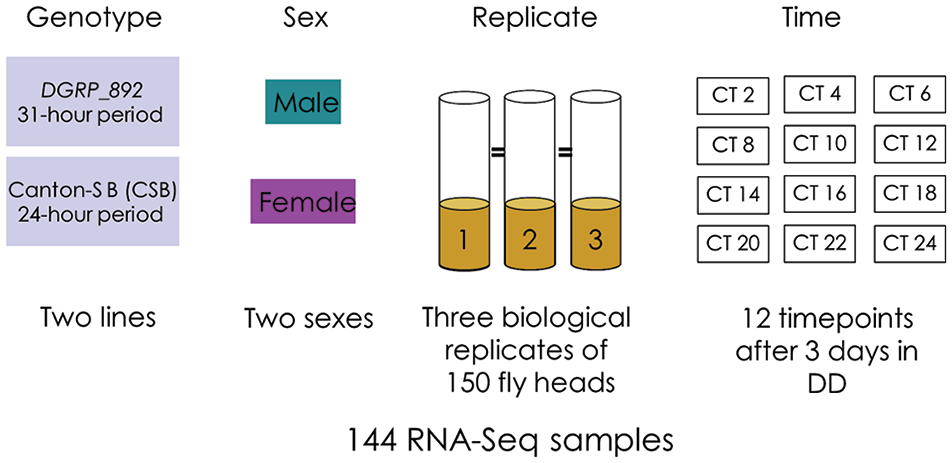
Experimental setup.
Numbers of differentially expressed genes by factor in the generalized linear model.
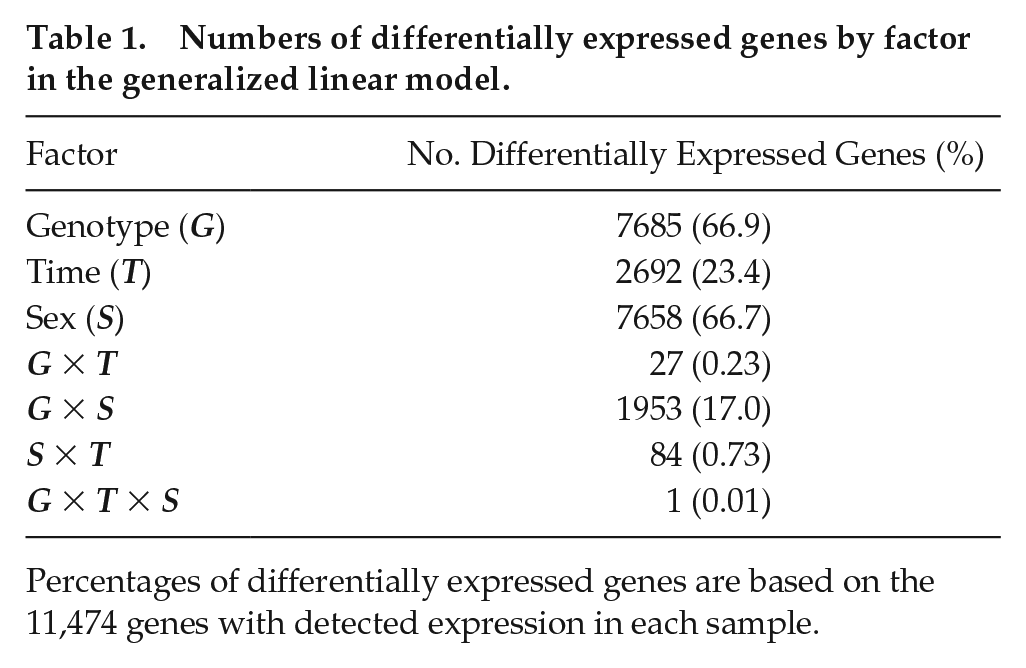
Percentages of differentially expressed genes are based on the 11,474 genes with detected expression in each sample.
Differential Expression Between DGRP_892 and CSB
Many of the 11,474 detectable genes were differentially expressed between DGRP_892 and CSB: 66.9% (7685) genes were differentially expressed by Genotype, similar to other studies which observed that 66% to 72% of the detected genes had differential gene expression among genotypes of the DGRP (Lin et al., 2016a; Everett et al., 2020). Some examples of genes with differential expression between the two genotypes are shown in Figure 2a. Slightly less than half (47.9%) of the transcripts had higher gene expression in DGRP_892 compared with CSB. The vast majority of these genes (94.8%) were protein coding genes, with the remaining classified as non-protein coding (Suppl. Table S2). We used MMC to cluster all 7685 significantly differentially expressed genes into modules (Stone and Ayroles, 2009; Suppl. Table S4). This procedure clustered genes with highly correlated expression into 15 large modules (Figure 2b). These modules predict co-regulated networks of gene expression (Harbison et al., 2009). Gene Ontology (GO) analyses (Yu et al., 2012) identified significant enrichment for many different terms in each module (Suppl. Table S5). For example, Module 2 was enriched for genes involved in circadian rhythms and sleep, such as Cul3, Hk, jet, Pka-R2, Rdl, Sh, and sgg (Figure 2b; Suppl. Table S5; Park et al., 2000; Martinek et al., 2001; Cirelli et al., 2005; Koh et al., 2006; Bushey et al., 2007; Parisky et al., 2008; Stavropoulos and Young, 2011). Module 2 contains a large group of genes (725 total) potentially interacting with these circadian rhythm/sleep genes. Thus, most of the genes with detectable expression in the head were differentially expressed among genotypes, and some of these genes included known circadian and sleep genes.
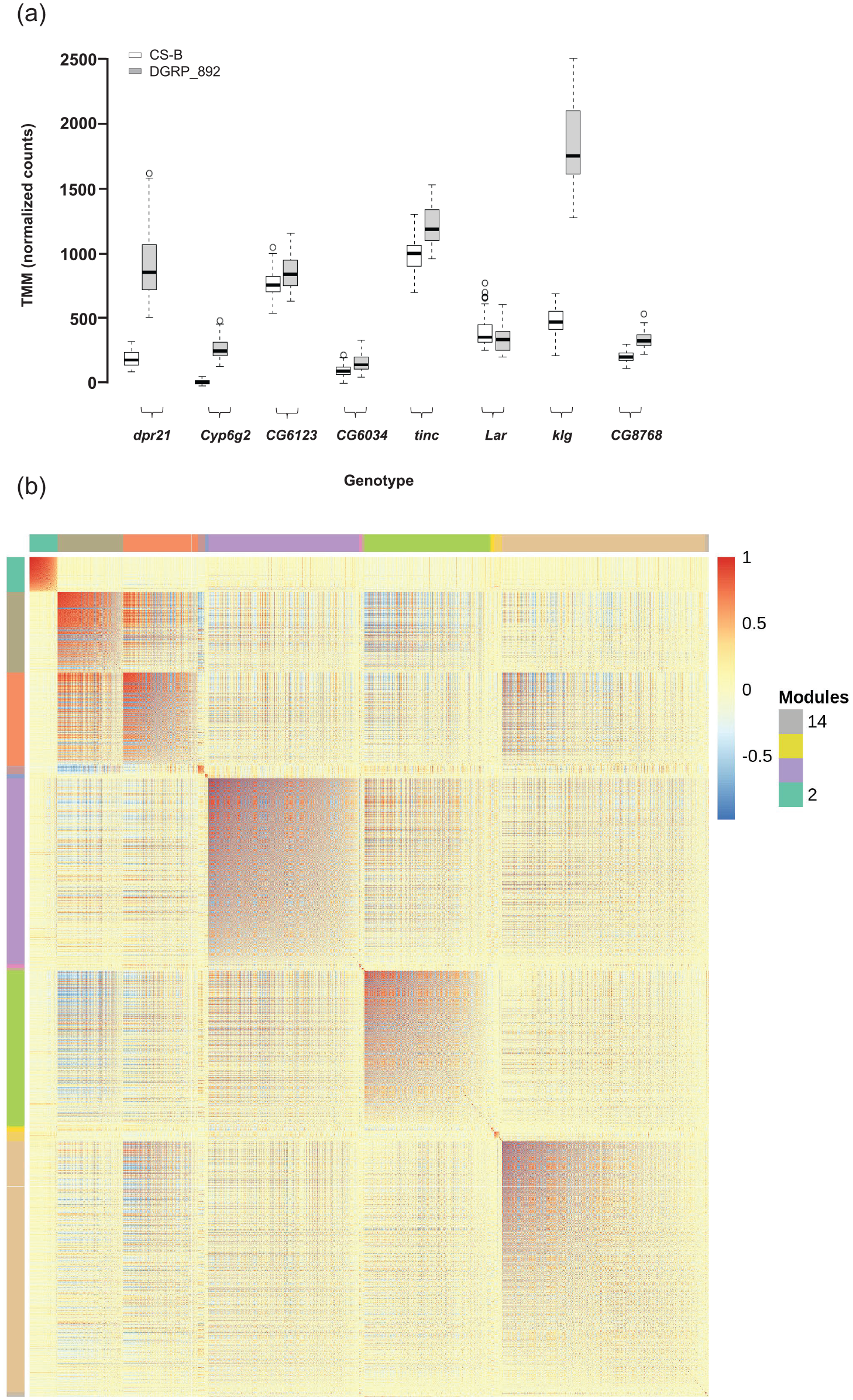
Genes differentially expressed between DGRP_892 and CSB. (a) Representative boxplots of TMM-normalized gene expression for dpr21, Cyp6g2, CG6123, CG6034, tinc, Lar, klg, and CG8768. (b) Plot of modules identified using Modulated Modularity Clustering. Each square in the plot represents the correlation between two genes; all 7685 genes are shown in the plot. The red-white-blue scale bar ranges high positive correlation (1.0, red) to high negative correlation (–1.0, blue). Colored bars demarcate the boundary of each module. Clusters of genes (modules) range from the most highly interconnected (upper left) to the least interconnected (lower right). Abbreviations: CSB = Canton-S B; TMM = trimmed mean of M-values.
We found that 166 of the 7685 differentially expressed genes overlapped with 268 previously identified in the GWAS of circadian behavior, an enrichment over what would be expected by chance (Suppl. Table S6; p < 0.0001, hypergeometric test). Interestingly, only 34 of the 166 genes were also significant for the main effect of Time or the Genotype × Time interaction, raising the possibility that some of the effects on circadian period occur in a time-independent fashion. We therefore chose candidate genes from the overlap of the two studies for further testing (see “Verification of Candidate Genes”).
Differential Gene Expression Across Time
Roughly one quarter of the genes with detectable expression also varied significantly by time point across 24 h (Table 1; Suppl. Table S2). Some representative examples of changes in gene expression over time can be seen in Figure 3a, including the canonical circadian clock genes per and Clk, whose transcripts are known to cycle over 24 h (Hardin et al., 1990; Hardin, 2004). We used MMC to cluster the gene expression of all 2692 genes. MMC analysis clustered the correlation among each gene pair into eight modules, including one very large module with 1637 genes, Module 4 (Figure 3b; Suppl. Table S7). Only Module 4 was enriched for GO terms (Suppl. Table S8). Genes that vary significantly with time may be cycling in a circadian fashion, or they may simply have differential gene expression at a single time point. We therefore used these 2692 genes in a further analysis to determine whether the time-specific differences in gene expression could be attributed to cycling behavior (see JTK_CYCLE and RAIN analyses below).
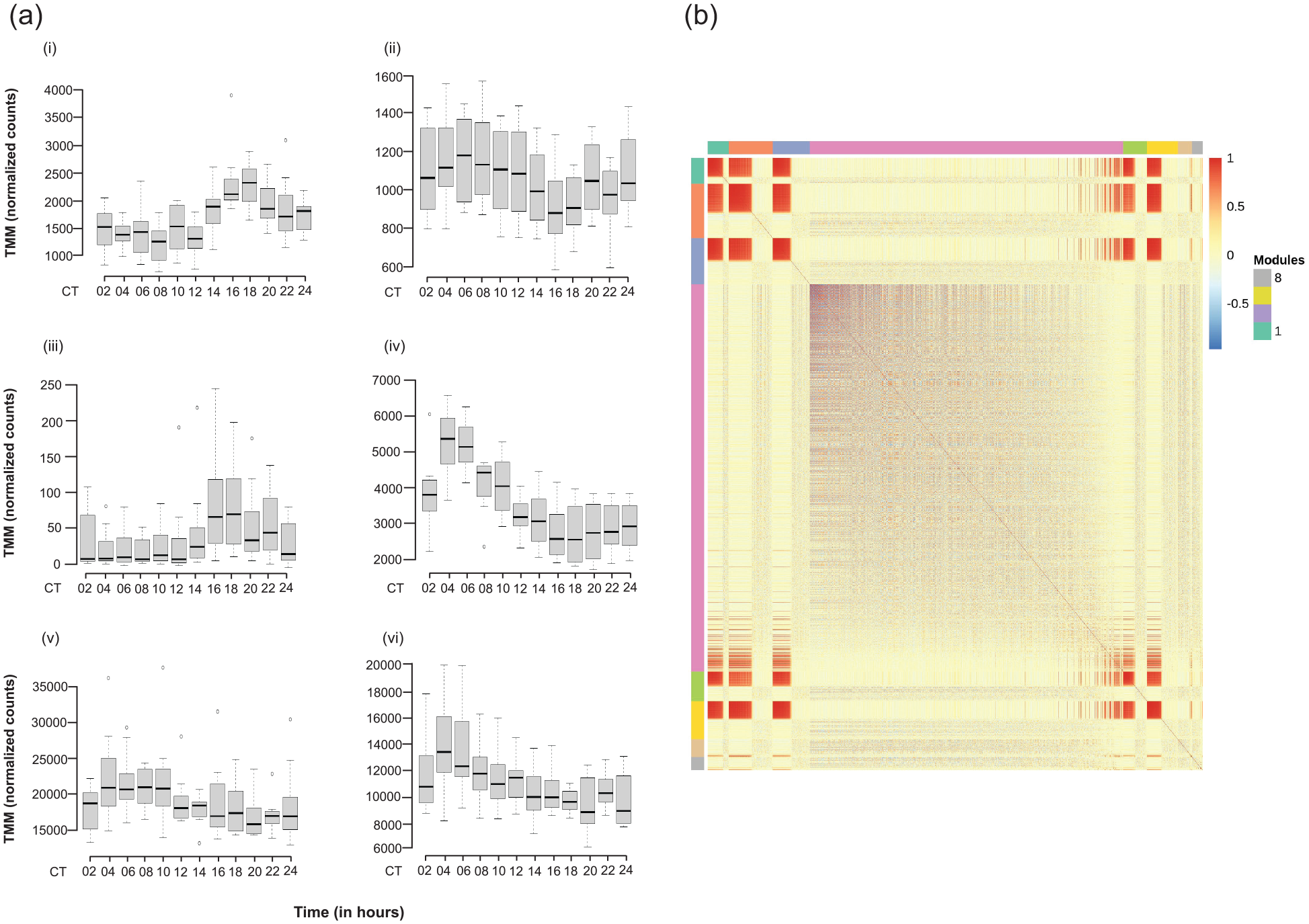
Genes differentially expressed with time. (a) Representative box plots of TMM-normalized gene expression for (i) per, (ii) Clk, (iii) dy, (iv) gol, (v) inaF-B, and (vi) Slob are plotted against time point. The box plots represent the distribution of TMM-normalized gene expression values for both genotypes and sexes combined. CT denotes circadian time in constant darkness. (b) Plot of modules identified using Modulated Modularity Clustering. Each square in the plot represents the correlation between 2 genes; all 2692 genes are shown in the plot. The red-white-blue scale bar ranges from high positive correlation (1.0, red) to high negative correlation (–1.0, blue). Colored bars demarcate the boundary of each module. Clusters of genes (modules) range from the most highly interconnected (upper left) to the least interconnected (lower right). Abbreviation: TMM = trimmed mean of M-values.
Significant Genotype × Time Differential Gene Expression
In addition to genes differentially expressed by the main effects of Genotype and Time, we wanted to know whether any of the genes were differentially expressed by the interaction of Genotype and Time. We hypothesized that if significant Genotype × Time interactions in gene expression existed, it would suggest potential differences in cycling behavior that might underlie the long circadian period. Only 27 genes had significant differential gene expression for this interaction term (Suppl. Table S2). per was the only canonical clock gene with a significant Genotype × Time interaction. Some representative genes have been plotted in Figure 4a. Using MMC, the pairwise correlation of gene expression among these 27 genes clustered into four modules (Suppl. Table S9). Due to the small number of genes significant for Genotype × Time, we did not observe much GO term enrichment in the modules, with the exception of Module 1 (Suppl. Table S10). This module consisted of three heat shock proteins, Hsp23, Hsp26, and Hsp27, which were associated with the GO terms protein folding/refolding and response to temperature and heat shock stress. As with the genes that were differentially expressed by Time, significant Genotype × Time terms might represent circadian cycling. These genes were also used in the analysis to identify cycling genes outlined below.
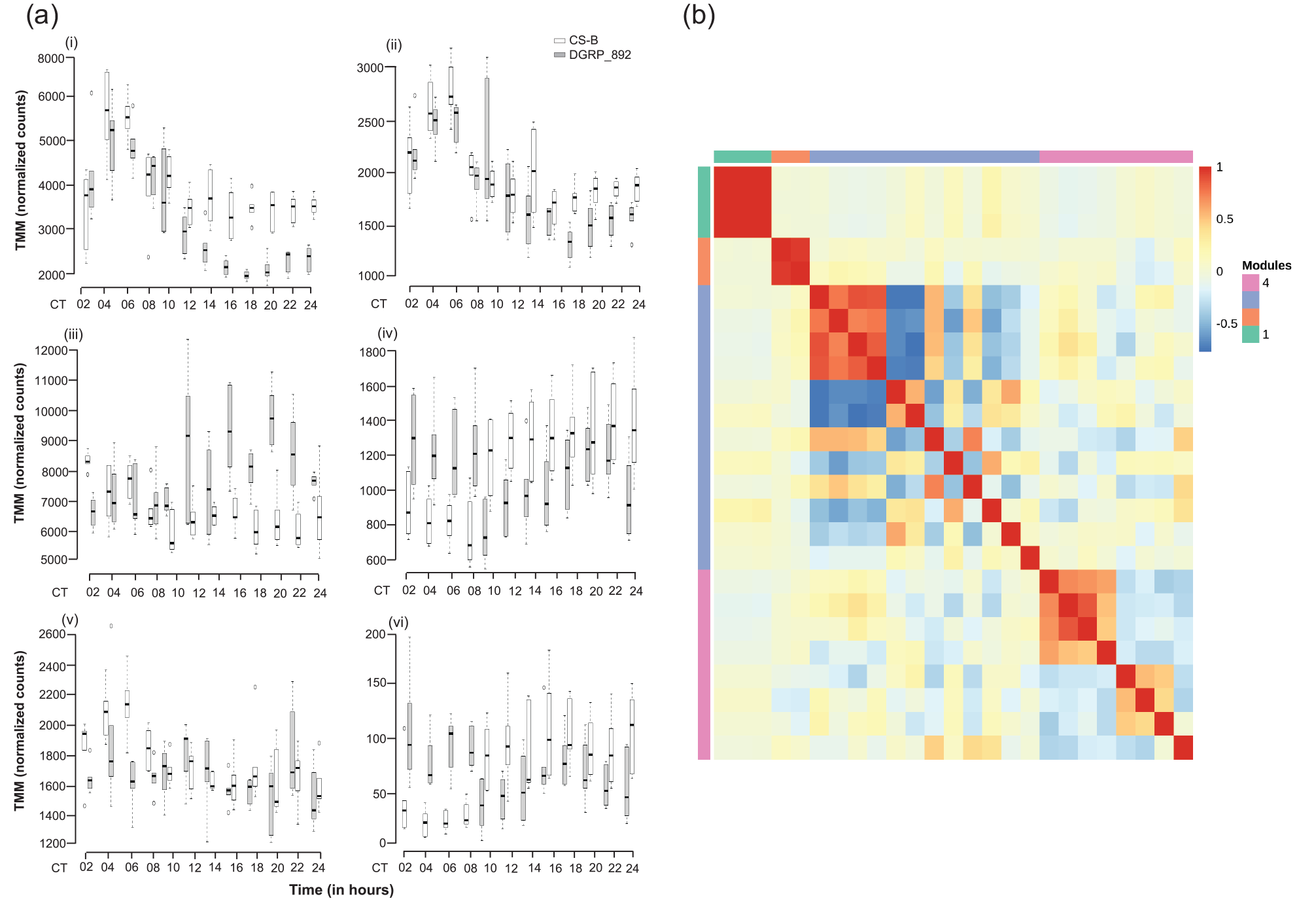
Genes with a significant Genotype × Time interaction. (a) Representative box plots of TMM-normalized gene expression for (i) gol, (ii) Usp8, (iii) Rh5, (iv) Cyp6a21, (v) Dnr1, and (vi) CG33965 are plotted against time point for DGRP_892 (gray boxes) and CSB (white boxes). CT denotes circadian time in constant darkness. (b) Plot of modules identified using Modulated Modularity Clustering. Each square in the plot represents the correlation between two genes; all 27 genes are shown in the plot. The red-white-blue scale bar shows ranges from high positive correlation (1.0, red) to high negative correlation (–1.0, blue). Colored bars demarcate the boundary of each module. Clusters of genes (modules) range from the most highly interconnected (upper left) to the least interconnected (lower right). Abbreviation: CSB = Canton-S B; TMM = trimmed mean of M-values.
Sex-specific Differential Gene Expression
Although both sexes had an elongated circadian period, a 50.4-min difference was observed between male and female flies of DGRP_892, with males having a longer circadian period than females (Harbison et al., 2019). Thus, sex-specific differences in gene expression may contribute differentially to the long period. Accordingly, we sequenced RNA from males and females separately. As anticipated from many previous experiments using Drosophila (Jin et al., 2001; Arbeitman et al., 2002; Parisi et al., 2003; Ranz et al., 2003; Harbison et al., 2005; Wayne et al., 2007; Zhang et al., 2007; Ayroles et al., 2009; Huylmans and Parsch, 2014; Huang et al., 2015; Lin et al., 2016a; Everett et al., 2020), most of the genome (66.7%) was differentially expressed between male and female heads (Table 1; Suppl. Table S2). We observed an enrichment of sex-specific differential expression on the X chromosome. The number of differentially expressed genes on the X chromosome, 1290, is 9.95% greater than would be expected by chance, while the number of differentially expressed genes on Chromosome 2L, 1422, is 7.62% lower than expected by chance (chi-square test, p = 0.001). MMC analysis clustered these genes into seven large modules with many significant GO terms (Suppl. Tables S11 and S12). Similarly, 1953 genes were differentially expressed by the Genotype × Sex interaction term, and 84 by the Sex × Time interaction term (Table 1). MMC clustering identified 13 modules for Genotype × Sex and 4 modules for Sex × Time (Suppl. Tables S13 and S14, respectively) and corresponding GO terms (Suppl. Tables S15 and S16, respectively). Only one gene was significant for the three-way interaction of Genotype × Time × Sex, Btk29A. Sex-specific differences in gene expression were ubiquitous in fly heads and may therefore contribute to sex-specific differences in the long circadian period.
JTK_CYCLE and RA IN Analysis to Identify Cycling Gene Expression
As mentioned previously, differences in gene expression across time may be due to circadian cycling, or they may be due to a difference in gene expression at a single time point (Hsu and Harmer, 2012). We used more robust analyses to determine whether the genes differentially expressed by Time or Genotype × Time were in fact cycling. We used JTK_CYCLE and RAIN analysis to predict whether these genes were cycling with a 24-h period (Hughes et al., 2010; Thaben and Westermark, 2014). We performed the analysis for each genotype and sex separately. Using JTK_CYCLE, 343 unique genes were predicted to cycle with 24-h periodicity (adjusted p < 0.05). RAIN analysis predicted 1286 unique genes would cycle with 24-h periodicity (adjusted p < 0.05), and all of the genes identified by JTK_CYCLE were also predicted by RAIN (Suppl. Table S17). Cycling transcripts were easier to detect in female heads than in males: Only 34.4% of the transcripts predicted to cycle came from the male data. As expected, most of the putatively cycling transcripts were identified from CSB, the line with the 24-h period; only 14.3% of the transcripts predicted to cycle came from DGRP_892 data. Overall, 132 genes were predicted to cycle in both DGRP_892 and CSB; 1102 were predicted to cycle in CSB only, and 52 were predicted to cycle in DGRP_892 only.
Canonical clock genes were predicted to cycle using these analyses. For CSB, per was the only canonical clock gene predicted to cycle in both sexes using JTK_Cycle, while Clk and vri were predicted to cycle in CSB in one sex only. None of the canonical clock genes were predicted to cycle in DGRP_892 by JTK_Cycle. When the less restrictive RAIN algorithm was used, however, canonical clock genes per, tim, and Clk were predicted to cycle in both genotypes, while Pdp1 and vri were predicted to cycle in CSB only.
In addition, we searched for genes putatively cycling at longer period lengths, from 26 to 32 h. Only one gene, gol, was predicted to cycle over a 28-h period in DGRP_892 using JTK_Cycle, and it was specifically predicted to cycle in females. Interestingly, gol was also predicted to cycle in both sexes of CSB with a 24-h period. We found that 24 of the 1286 genes predicted to cycle by RAIN overlapped with the 268 identified in the previous GWAS, including gol (Harbison et al., 2019). We subjected a subset of these genes to further verification and testing as outlined below.
Verification of Candidate Genes
As mentioned previously, 166 genes significant for the main effect of Genotype overlapped with the GWAS. Among these 166 genes, 24 genes were predicted to cycle from the JTK_CYCLE/RAIN analysis, and 39 are expressed in clock neurons (Kula-Eversole et al., 2010; Nagoshi et al., 2010; Abruzzi et al., 2017; Suppl. Table S18). We identified 45 Minos insertion mutant lines available from these 166 genes to test for their effects on circadian period. Ten of these genes were predicted to cycle in CSB and not DGRP_892. The remaining 35 lines came from the overlap of Genotype/GWAS. Initially, 17 mutants exhibited significant effects on circadian period. We repeated the phenotypic measures and found that 11 mutant alleles had significant effects on circadian period that were confirmed by re-test: Alk, alph, CG10089, CG42540, CG6034, CG6123, CG8768, klg, Lar, sick, and tinc (Bonferroni corrected p value = 0.001; Suppl. Table S1). Interestingly, the effect of the mutation in Alk was largely confined to females, while the effect of CG10089 was predominantly due to a small shift in male behavior (Suppl. Table S1). In addition, CG6123 was reported to have an effect on circadian period previously (Harbison et al., 2019). Minos insertions in CG6123, klg, and tinc had the greatest increases in circadian period over the control, increasing period by 1.63, 1.70, and 0.97 h on average in both males and females, respectively (Suppl. Table S1). Thus, we identified genes altering circadian period, including some mutants that produced large increases in period when tested.
Genetic Rescue of CG6123
We generated a precise excision of the Minos element in CG6123 (see “Materials and Methods”). We verified the excision through PCR and sequencing analysis (Suppl. Figs. S2-S4). We measured the locomotor activity rhythms and compared the circadian period of the precise excision line with the original homozygous CG6123 Minos element line, a heterozygous cross between the CG6123 Minos element line and the excision line, and the w1118 control line. As Figure 5 shows, both the homozygous excision line and the heterozygous cross were indistinguishable from the w1118 control line. This demonstrates genetic rescue of the long-period phenotype, and the effects of the rescued allele are dominant to the mutant allele. We have renamed CG6123 Kairos, a Greek term which refers to an appropriate or suitable moment for action.
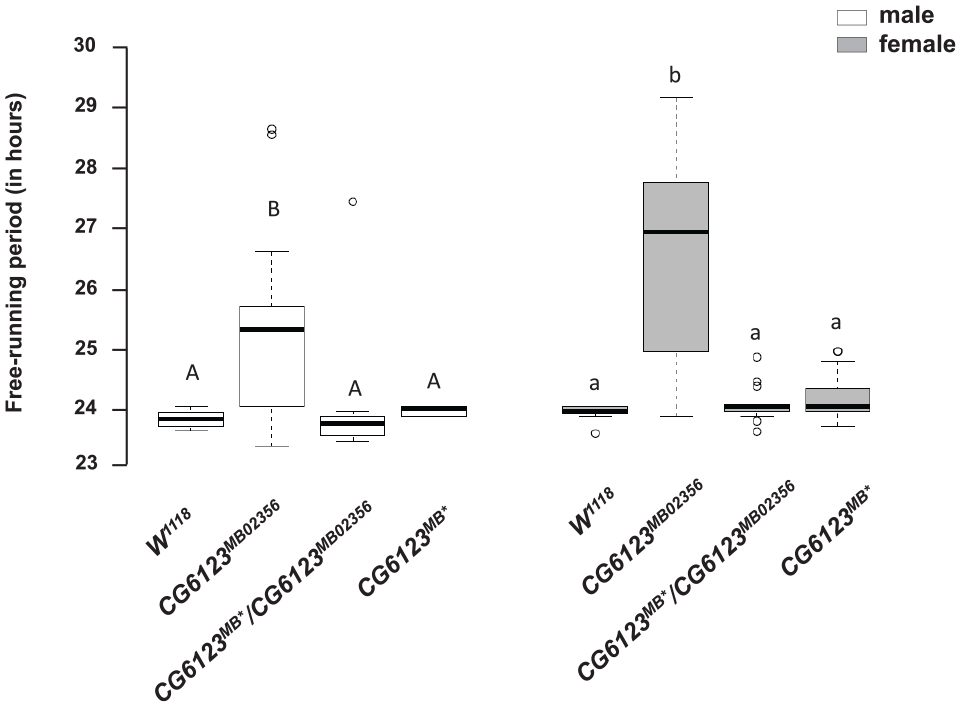
Genetic rescue of the Minos element in Kairos (CG6123). Free-running period of the w1118 isogenic control, homozygous CG6123MB02356 mutant, CG6123MB02356/CG6123MB* (heterozygote) and the homozygous CG6123MB* precise excision under constant dark conditions. White boxes indicate males and gray boxes indicate females. N = 16 in each case except for CG6123MB* females where N = 15. Letters above the boxplots indicate significant differences by post hoc Tukey test (p < 0.05).
Discussion
In the present study, we have surveyed the transcriptome of a wild-type Drosophila line, DGRP_892, which exhibits an extremely long period in its locomotor activity behavior in constant darkness. Our approach to the experimental design followed the “Guidelines for genome-scale analysis of biological rhythms” (Hughes et al., 2017) and included (1) sample collection every 2 h, (2) biological replicates (there are six samples for every time point for each genotype), (3) greater than ten million reads per sample for flies (14,638,141 on average per sample for our data), (4) pooling of five or more individuals (we used 150 heads per sample), (5) samples collected in constant darkness and under constant temperature, and (6) samples from both sexes were collected. The only exception to the guidelines in our experiment was the collection of samples over two circadian cycles (i.e., 48 h); instead, we collected samples over 24 h only. As the guidelines state, this lack of duplicate time points could potentially lead to false negatives when attempting to identify cycling genes (Hughes et al., 2017). per was the only canonical clock gene with a significant Genotype × Time interaction term. per was also the only canonical clock gene predicted to cycle for both sexes in CSB using JTK_Cycle, while Clk and vri were predicted to cycle for one sex only. However, all of the canonical clock genes (per, tim, Pdp1, Clk, and vri) were predicted to cycle in CSB using the less stringent algorithm in RAIN. In addition, cycling transcripts are more easily detected when clock neurons are assayed separately as opposed to extracting RNA from the entire head (Kula-Eversole et al., 2010). These results suggest that clock gene expression measured using RNA-Seq techniques is relatively noisy and cycling transcripts may evade detection unless additional time points are sampled, more permissive algorithms are used, or smaller subsets of tissue are sampled.
Fewer oscillating transcripts were present in DGRP_892 than CSB. Canonical clock genes did not appear to cycle in DGRP_892 when the data were analyzed using JTK_Cycle; however, per, tim, and Clk were predicted to cycle with 24-h periodicity when the more permissive RAIN algorithm was used. These contradictory findings suggest two distinct possibilities. The first is that per, tim, and Clk are cycling in 24 h, and the gene(s) contributing to the long circadian period act downstream of the molecular clock. The second possibility is that per, tim, and Clk are not cycling in DGRP_892 and that the identification of these genes as cycling by RAIN is a false positive result. If the genes are not cycling in DGRP_892, then the potential exists for at least one of the genes contributing to the long circadian period to interact directly with the molecular clock. We previously measured gene expression in constant darkness in per, tim, and Pdp1 in DGRP_892 using real-time PCR (RT-PCR) and analyzed the data using JTK_Cycle; none of the genes cycled (Harbison et al., 2019), consistent with the second possibility.
We found transcripts that have not been shown to cycle in previous studies (Hughes et al., 2012; Rodriguez et al., 2013). We compared the total number of putative oscillating genes across previous expression studies, and found that 10 to 35 genes overlap (Suppl. Table S19; Claridge-Chang et al., 2001; McDonald and Rosbash, 2001; Ceriani et al., 2002; Lin et al., 2002; Ueda et al., 2002; Keegan et al., 2007). The relatively low overlap of cycling genes between these previous studies and ours could have several origins, including (1) the genotypes assayed, (2) the platform used to assess gene expression (RNA-Seq vs. microarray), (3) the number of replicated samples, (4) the number of time points assayed, and (5) the algorithms used to identify cycling genes.
Approximately two thirds of all detected transcripts (out of 11,474) were differentially expressed between genotypes while only 27 were differentially expressed for the Genotype × Time interaction, which suggests that at least some of the effects of gene expression on circadian period may be due to time-independent gene activity. Combining GWAS (Harbison et al., 2019) and RNA-Seq analysis, we were able to narrow down candidate genes that might be critical for regulating circadian rhythms of the locomotor activity behavior. Further testing confirmed 11 genes affecting circadian period for the locomotor activity rhythms. Genetic rescue revealed that Kairos (CG6123) is necessary for regulating free-running locomotor activity behavior. Our RNA-Seq data also revealed that Kairos does not cycle in constant darkness. The molecular function of Kairos is unknown. It is conserved among Diptera and other insects, but does not have a known mammalian ortholog (Hu et al., 2011). It is highly enriched in the ventral lateral neurons (LNvs; Abruzzi et al., 2017), particularly the small ventral lateral neurons (sLNvs; Kula-Eversole et al., 2010), suggesting a potential role as an input molecule to the core clock. Further experiments will determine whether it interacts directly with canonical circadian clock genes.
In addition to Kairos, we also verified that Alk, alph, CG10089, CG42540, CG6034, CG8768, klg, Lar, sick, and tinc alter the period lengths of rest: activity behavior. The Minos insertion mutants of klg in particular displayed a comparatively longer period than the rest. klg encodes a cell-adhesion molecule expressed in both neurons and glia and is required for long-term memory formation (Matsuno et al., 2015). klg expression has been localized to the sLNvs (Kula-Eversole et al., 2010). klg regulates the transcriptional activator repo in glial cells (Matsuno et al., 2015). It would be interesting to investigate the role of klg in the regulation of circadian rhythms given the importance of glial cells in rhythmic behavior (Ng et al., 2011).
The candidate genes sick, tinc, CG10089, CG42540, CG6034, and CG8768 have not been previously reported for their role in circadian regulation in flies, nor have their mammalian homologs. sick is expressed in dorsal neurons (DN1) as well as large and small LNvs (Kula-Eversole et al., 2010; Abruzzi et al., 2017). sick encodes a cytoskeleton protein and is highly expressed in F-actin rich axons of mushroom bodies (Abe et al., 2014). Accordingly, sick mutants exhibit axonal growth defects in the mushroom bodies (Abe et al., 2014). Interestingly, knockdown of sick in the dopaminergic neurons enhances olfactory memory, which suggests that the normal function of sick is to act as a memory suppressor gene (Walkinshaw et al., 2015). tinc is highly enriched in central and peripheral nervous system and encodes a transmembrane protein, which is involved in development of the compound eyes in D. melanogaster (Hirota et al., 2005). CG10089 is expressed in both large and small LNvs (Kula-Eversole et al., 2010). Electronic annotation suggests that CG10089 has protein tyrosine/serine/threonine phosphatase activity, but little else is known about this gene (dos Santos et al., 2015). Likewise, CG42540 has an unknown function but exhibits some homology to human stomatin; CG6034 is predicted to have oxidoreductase activity; and CG8768 is predicted to have catalytic activity (dos Santos et al., 2015). Further investigation is needed to elucidate the role of these genes in modifying the circadian period of locomotor activity behavior.
Several of the genes we verified have been noted in previous circadian and sleep studies. Lar is a gene implicated in Drosophila rhythmic behavior in constant darkness (Agrawal and Hardin, 2016) and in environmental sensitivity to waking activity (Harbison et al., 2013). Lar encodes a receptor protein tyrosine phosphatase leukocyte-antigen-related protein, and Lar mutants lack the dorsal projections of core clock neurons, which renders flies completely arrhythmic in constant darkness (Agrawal and Hardin, 2016). The arrhythmic phenotype of Lar mutants was largely attributed to impairment of pigment-dispersing factor (PDF) accumulation in the core clock neurons instead of effects on the core clock gene expression levels (Agrawal and Hardin, 2016). These results are in contrast with our results as the Minos insertion mutants of Lar that we tested are completely rhythmic. One possibility is that the Lar mutants used in our study act as a hypomorph rather than a deletion as the Minos insertion is in an intron. Other possibilities may include differences in the genetic background, the external environment, and their interaction, which are known to influence expressivity and penetrance differentially (Chandler et al., 2017). Alk encodes Anaplastic lymphoma kinase, which is involved in sleep regulation and memory formation (Bai and Sehgal, 2015). Mutants of Alk have increased sleep when its expression levels are reduced in the mushroom bodies (Bai and Sehgal, 2015). Interestingly, previous work found that Alk does not affect locomotor circadian behavior, although it is known to cycle in dorsal lateral neurons (LNds; Bai and Sehgal, 2015; Abruzzi et al., 2017). However, only males were tested for circadian behavior (Bai and Sehgal, 2015). Our test, which examined both male and female mutants of Alk, revealed an effect on circadian period predominantly in females, who had a 43-min decrease in circadian period compared with the control (Suppl. Table S1). alph has been shown to cycle in constant darkness in an earlier microarray study, but did not cycle in LD (Ueda et al., 2002). We did not observe significant cycling for alph. alph encodes a Serine-Threonine phosphatase involved in RAS/MAPK signaling (Baril and Therrien, 2006). Thus, several of the genes we identified have known roles in circadian rhythms and sleep.
Systems genetics approaches combine the results of large-scale measures of the genome, transcriptome, proteome, and so on, to identify the most promising candidate genes influencing a complex trait (Civelek and Lusis, 2014). Our combinatorial analysis of transcriptomic data together with GWAS results enabled us to identify genes that may underlie the long-period phenotype seen in DGRP_892. Of the 11 mutants with significant effects on circadian period, none precisely phenocopy the 31-h circadian period of DGRP_892. This strongly indicates that multiple genes are interacting to produce the long period, a notion supported by work reporting the interaction of naturally occurring variants to affect rhythmic behavior in flies (Peschel et al., 2006). Using CRISPR, it is possible to perturb up to four genes simultaneously with a single polycistronic construct (Port and Bullock, 2016). The application of CRISPR-Cas9 tools will enable us to perturb multiple combinations of genes in DGRP_892 to understand how putative genetic interactions produce the long circadian period. It is also possible that some of the candidate genes may have effects that are developmental in origin. Perturbation of candidate genes using conditional GAL4 systems (Osterwalder et al., 2001; McGuire et al., 2003) will reveal any temporal dependency. Genes can be placed into their proper genomic context by verifying the transcriptional changes among associated genes common to a given module. For example, perturbations made to Alk in the DGRP_892 background should lead to a corresponding transcriptional response in the circadian/sleep genes highly correlated with Alk from the same module (Harbison et al., 2009). In this way, the contributing effectors producing the long period can be placed among the known circadian and sleep network.
Supplemental Material
sj-pdf-1-jbr-10.1177_0748730420975946 – Supplemental material for Identification of Genes Contributing to a Long Circadian Period in Drosophila Melanogaster
Supplemental material, sj-pdf-1-jbr-10.1177_0748730420975946 for Identification of Genes Contributing to a Long Circadian Period in Drosophila Melanogaster by Shailesh Kumar, Ilker Tunc, Terry R. Tansey, Mehdi Pirooznia and Susan T. Harbison in Journal of Biological Rhythms
Supplemental Material
sj-xlsx-2-jbr-10.1177_0748730420975946 – Supplemental material for Identification of Genes Contributing to a Long Circadian Period in Drosophila Melanogaster
Supplemental material, sj-xlsx-2-jbr-10.1177_0748730420975946 for Identification of Genes Contributing to a Long Circadian Period in Drosophila Melanogaster by Shailesh Kumar, Ilker Tunc, Terry R. Tansey, Mehdi Pirooznia and Susan T. Harbison in Journal of Biological Rhythms
Footnotes
Acknowledgements
We thank the National Intramural Sequencing Center Consortium for the RNA sequences. This work used the computational resources of the NIH HPC Biowulf cluster (![]() ). The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Intramural Research Program of the NIH, the National Heart Lung and Blood Institute.
). The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Intramural Research Program of the NIH, the National Heart Lung and Blood Institute.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.