
Abstract
Caffeine is widely used to reduce sedation and increase alertness. However, long-term caffeine use may disrupt circadian (daily, 24-h) rhythms and thereby negatively affect health. Here, we examined the effect of caffeine on photic regulation of circadian activity rhythms in mice. We found that entrainment to a standard 12-h light, 12-h dark (LD) photocycle was delayed during oral self-administration of caffeine. Both acute, high-dose caffeine and chronic, oral caffeine exposure potentiated photic phase-delays in mice, suggesting a possible mechanism by which entrainment to LD was delayed. The effect of caffeine on photic phase-resetting was mimicked by administration of adenosine A1, but not A2A, receptor antagonist in mice. Our results support the hypothesis that caffeine interferes with the ability of the circadian clock to respond normally to light.
Caffeine is the most widely used psychoactive substance in the world. Although it is known for its ability to reduce sedation and increase alertness, habitual caffeine use has been reported to impair sleep and cause rebound sleepiness that reinforces caffeine intake for its alerting properties (Roehrs and Roth, 2008). However, tolerance to the stimulant effects of caffeine develops rapidly, and recent studies suggest that caffeine-induced disruption of circadian (daily, 24-h) rhythms may be important for its behavioral effects.
In mammals, circadian rhythms are regulated by the suprachiasmatic nucleus (SCN) of the hypothalamus, a clock whose timing, or phase, is entrained to the environment by photic (light) and nonphotic (behavioral) stimuli (Moore, 1983; Pickard, 1982; Webb et al., 2014). Photic input is mediated by glutamate release into the SCN from retinal afferents (Ding et al., 1997; Moore, 1983; Pickard, 1982), while nonphotic input is mediated primarily by serotonergic afferents from the median raphe nucleus (Glass et al., 2003). Interestingly, adenosine also appears to play a role in regulating SCN clock timing. Activation of presynaptic adenosine A1 receptors on retinohypothalamic tract neurons inhibits glutamate release into the SCN and thereby attenuates photic phase-resetting (Elliott et al., 2001; Hallworth et al., 2002; Sigworth and Rea, 2003; Watanabe et al., 1996). Conversely, adenosine A1 receptor agonist CPA mimics behavioral phase-shifts (Antle et al., 2001), consistent with the known opposing regulation of nonphotic stimuli on responses to photic input, and vice versa.
Caffeine is an adenosine A1/A2A receptor antagonist, so it is not surprising that it has been shown to alter circadian rhythms. Caffeine lengthens free-running (endogenous) circadian period in human cells and in rodents (Burke et al., 2015; Oike et al., 2011; van Diepen et al., 2014). While it does not induce circadian phase-shifts on its own at midday, caffeine attenuates sleep deprivation-induced nonphotic phase-resetting in hamsters (Antle et al., 2001) and increases light responsiveness of the murine SCN (van Diepen et al., 2014). A recent study in humans showed that caffeine intake in the late evening delayed melatonin rhythms and implicated A1 receptor inhibition in the mechanism of action (Burke et al., 2015). Here, we tested the hypothesis that caffeine would alter light-entrained activity rhythms and potentiate photic phase-delays in mice.
Methods
Animals
Adult (age 8-10 weeks at start of experimentation), male C57BL/6J mice were maintained in a 12-h light/12-h dark (LD) photocycle (except as noted below), individually caged, and their general circadian locomotor activity rhythms were monitored using infrared motion detectors interfaced with a computerized data acquisition system (ClockLab; Coulbourn Instruments, Whitehall, PA). The intensity of daily light exposure was ~400 lux and spectrum was 4100 K (Sylvania Octron 800 XP Ecologic 3), and the ambient temperature was 22 °C. Food was available ad libitum throughout the experiments, as was water (except as noted below). Six separate cohorts of mice were tested as noted below. Animal care/handling and experimental procedures were approved by the Indiana University of Pennsylvania IACUC according to NIH guidelines.
Effect of Chronic Caffeine on LD Entrainment
General circadian locomotor activity rhythms for each individually housed mouse were monitored as described above. Once the mice from cohort 1 (n = 18) showed stable entrainment to the LD photocycle for at least 14 days, their water bottle was replaced with a caffeine solution (1.0 mg/mL in water) (Costa et al., 2008) for the next 14 days. All data were analyzed using the ClockLab circadian toolbox (Actimetrics, Wilmette, IL) for MATLAB (Mathworks, Natick, MA). Circadian period (tau) was determined using ClockLab’s chi-square periodogram analysis. The peak magnitude (amplitude) of the periodogram was used to estimate the robustness of the rhythm. Activity onset was defined as the first 10 min of activity that (1) exceeded 10% maximum daily rate, (2) was preceded by 4 h or more of inactivity, and (3) was followed by 30 min or more of sustained activity. Onsets were then calculated relative to zeitgeber time 12 (ZT12), the beginning of the dark phase (active phase for nocturnal rodents). Activity offset was defined as the final 10 min that was preceded by 60 min or more of sustained activity and followed by 4 h or more of inactivity and was calculated in relation to ZT0, the time of lights-on (inactive/rest phase for nocturnal rodents). Alpha (the duration of the active phase) was calculated as the period between activity onset and activity offset (hours).
Effect of Chronic Caffeine on Photic Phase-resetting
A separate cohort of mice (cohort 2) was used to examine the effect of chronic caffeine on photic phase-resetting. Once the mice showed stable entrainment to the LD photocycle, they were given access to caffeine in their drinking water as described above (n = 11) or were maintained on water (n = 11) for a total of 28 days. The first 14 days on caffeine (or water drinking controls) were used to determine baseline onsets. On day 15, a 30-min phase-delaying light pulse (25 lux) was delivered at ZT14 (2 h after lights-off), during which mice were monitored to ensure that they were awake with their eyes open. After the light pulse, mice were released into constant darkness (DD) for 14 days to assess phase-resetting responses (Aschoff, 1965). Caffeine drinkers were maintained on caffeine during this time to avoid the possibility of testing the effects of caffeine withdrawal as opposed to caffeine itself. Phase-shifts were calculated as the difference between the projected times of activity onset (defined as above) on the day after stimulation as determined by (1) back extrapolation of the least-squares line through activity onsets on days 2 through 10 after treatment and (2) extrapolation of the least-squares line calculated from activity onset data collected during the 10 days before treatment.
Effect of Acute Caffeine on Photic Phase-resetting
To rule out the possibility that effects attributed to caffeine intake would not reflect physiological tolerance to caffeine, we examined the effect of acute, single doses of caffeine in caffeine-naïve mice. To assess the effects of acute caffeine on photic phase-delays, we performed a separate set of experiments in two additional cohorts of caffeine-naïve mice (cohorts 3 and 4). Once mice showed stable entrainment to the LD photocycle for at least 14 days, they were divided into treatment groups. On the day of the experiment, mice in the first cohort received an intraperitoneal injection of either caffeine (3.0 mg/kg) (Hilbert et al., 2013) or equal volume saline (n = 11 per treatment), while mice in the second cohort received either caffeine (15.0 mg/kg) (Hilbert et al., 2013) or equal volume saline (n = 11 per treatment). Injections were undertaken in dim red light 15 min prior to the 30-min light pulse at ZT14. Mice were then released into DD for 14 days, and phase-resetting responses were measured as above.
To rule out the possibility that caffeine may have a phase-resetting effect of its own at ZT14, we performed a no-light pulse control experiment in a fifth cohort of mice. Once mice showed stable entrainment to the LD photocycle for at least 14 days, they were divided into treatment groups. On the day of the experiment, we administered caffeine (15.0 g/kg, i.p.; n = 11) or saline (n = 10) in dim red light 15 min before ZT14 but did not deliver a light pulse. Mice were then released into DD for 14 days, and phase-resetting responses were measured as described.
Effect of Selective Adenosine Receptor Antagonism on Photic Phase-resetting
Caffeine acts as an antagonist to both adenosine A1 and A2A receptors. Therefore, we undertook another experiment in a sixth cohort of mice to determine which receptor likely mediates the effect of caffeine on photic phase-resetting. Once mice showed stable entrainment to the LD photocycle for at least 14 days, they received either A1 receptor antagonist DPCPX (6.0 mg/kg), A2A receptor antagonist ZM-241385 (20.0 mg/kg), or vehicle (DMSO; n = 6-8 per treatment) in dim red light 30 min prior to a phase-delaying light pulse at ZT14. Mice were then released into DD to assess phase-shifts.
Statistical Analyses
LD entrainment measures were analyzed using paired Student’s t tests to find differences between baseline and caffeine exposure. Phase-resetting data were analyzed using either unpaired Student’s t tests or one-way ANOVA, depending on the number of treatment groups in each experiment. Results were considered significant when p < 0.05. Data are expressed as mean ± SEM.
Results
Caffeine Drinking Alters LD Entrainment
Caffeine consumption had a delaying effect on dark phase activity in mice (n = 18 for all measures), delaying activity onsets from a baseline of −26.04 ± 4.04 min relative to ZT12, the time of lights-off, to −101.50 ± 13.34 min (t = 6.174, p < 0.0001) (Fig. 1A). Activity offsets relative to ZT0, the time of lights-on, were also delayed (t = 4.972, p = 0.0001) (Fig. 1B), from a baseline average of −16.95 ± 5.16 min to −57.50 ± 7.78 min (negative numbers indicate that onsets and offsets occurred after, as opposed to before, ZT12 and ZT0, respectively). Surprisingly, caffeine drinking reduced alpha, the duration of the active phase (t = 2.583, p = 0.0193) (Fig. 1C), from 11.85 ± 0.14 h to 11.27 ± 0.26 h. Also interesting, caffeine reduced both the duration (t = 3.294, p = 0.0043) (Fig. 1D) and the intensity (t = 2.761, p = 0.0133) (Fig. 1E) of activity during the light phase (rest phase for nocturnal mice). Average light phase activity duration changed from a baseline of 14.99 ± 1.58 min to 9.87 ± 1.34 min during caffeine exposure, while light phase activity intensity averaged 62.88 ± 6.37 counts at baseline and 45.15 ± 6.25 counts during caffeine drinking. Neither the duration (t = 0.5222, p = 0.6082) (Fig. 1F) nor the intensity (t = 0.9542, p = 0.3534) (Fig. 1G) of activity during the dark phase was affected by caffeine consumption. Baseline dark phase activity duration was 372.50 ± 31.45 min compared with 383.80 ± 41.19 min during caffeine consumption, and baseline dark phase activity intensity was 1904.00 ± 214.60 counts compared with 2081.00 ± 298.40 counts during caffeine exposure. Representative actograms from a water drinker and a caffeine drinker in this experiment are shown in Figure 2.
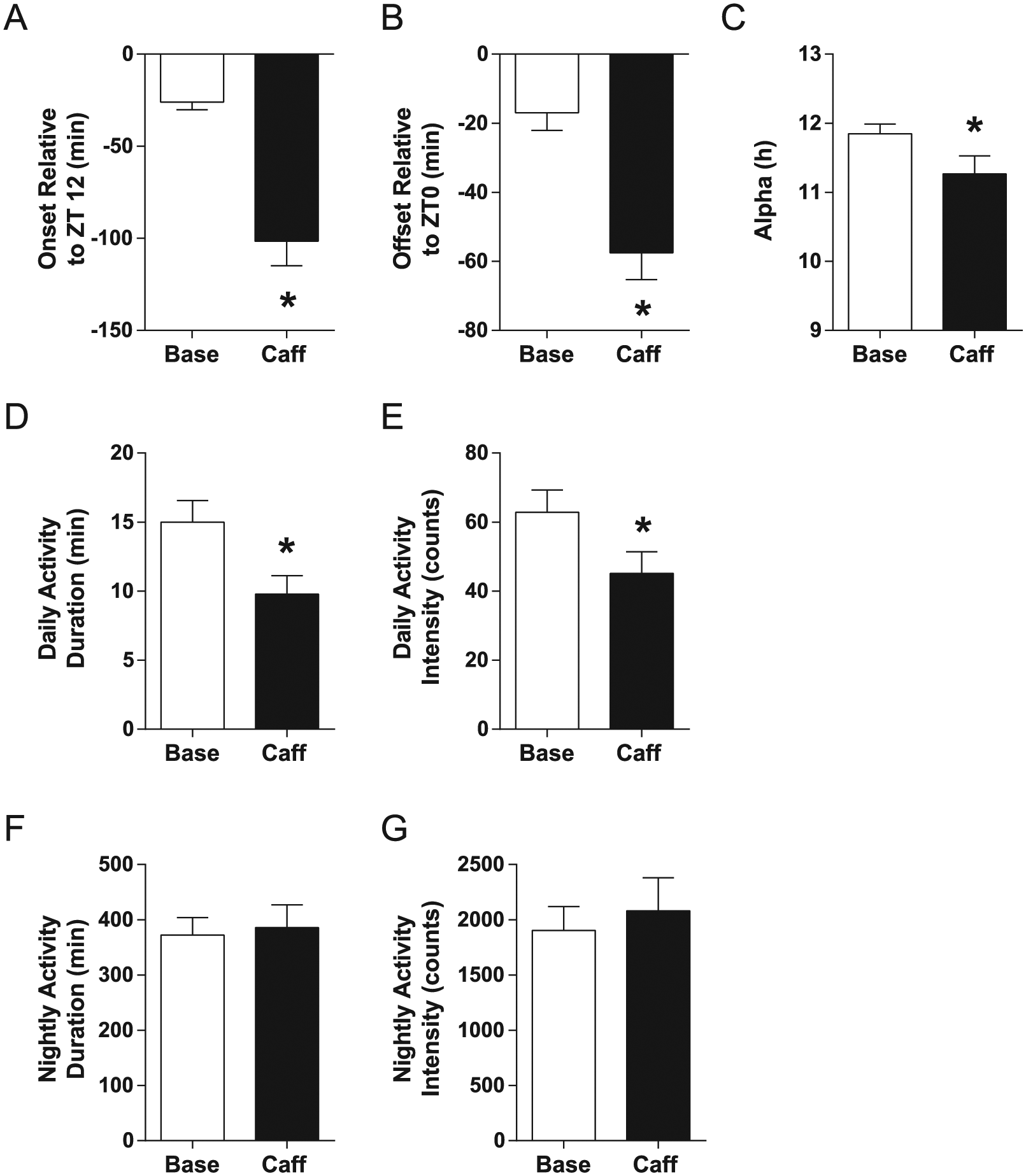
Caffeine delays light-entrained activity in mice. Two weeks of caffeine drinking (average = 102.40 ± 4.41 mg/kg/day) delayed both (A) activity onset and (B) offset and slightly but significantly reduced (C) active phase duration as well as (D, E) light phase activity duration and intensity. (F, G) No effect of caffeine consumption on dark phase activity was observed. Bars show mean ± SEM, *p < 0.05.
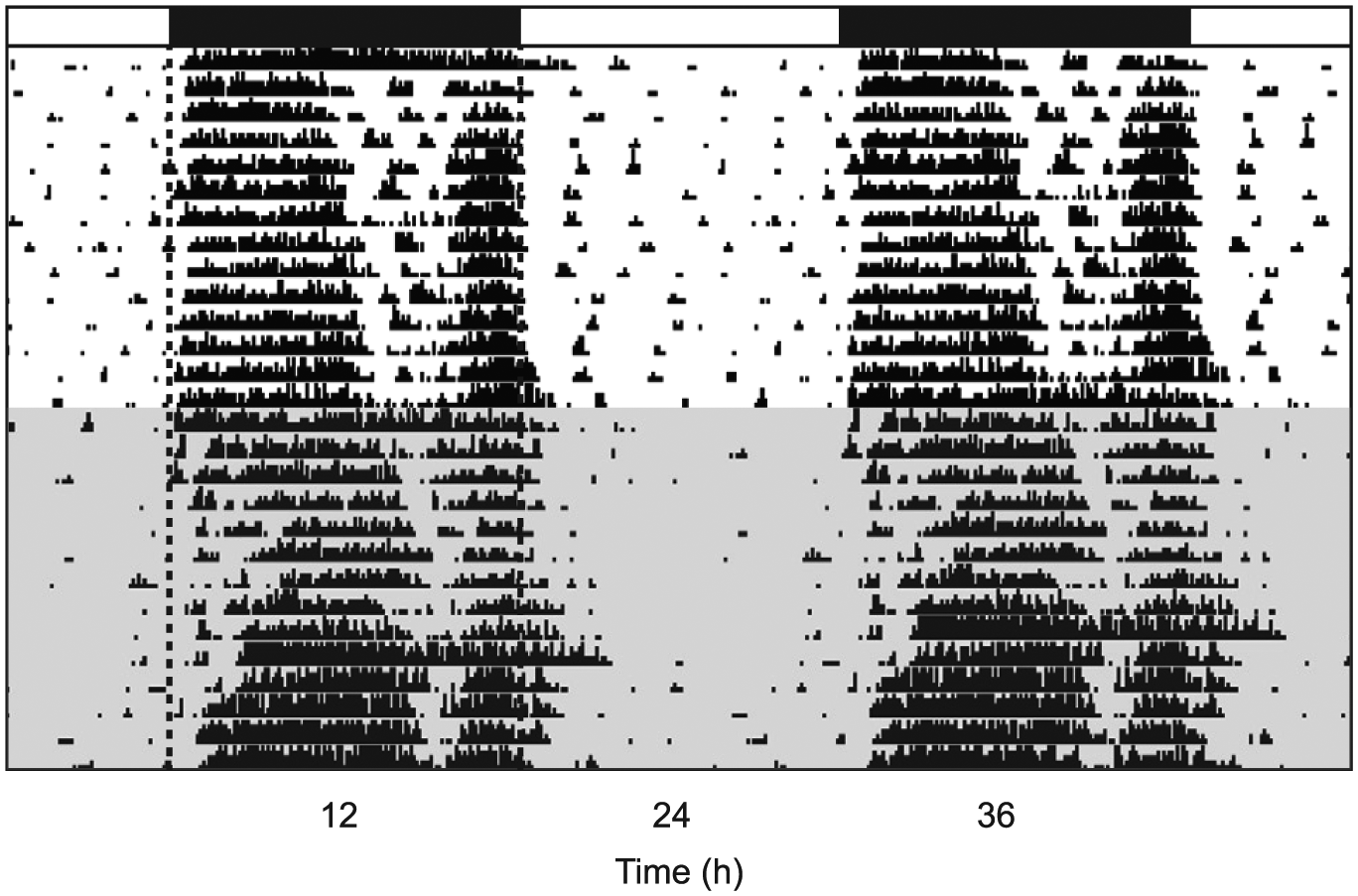
Representative, double-plotted actogram showing delayed light-entrained activity and reduced daytime activity in a mouse during caffeine consumption (shaded portion) compared with baseline measures (unshaded portion). Black bars denote the dark phase; dashed line added to the beginning and end of the dark phase for clarity.
Caffeine Drinking Enhances Photic Phase-Delays
Caffeine consumption potentiated photic phase-delays in mice relative to those measured in water-drinking controls (t = 3.25, p = 0.004) (Fig. 3), with caffeine drinkers showing 1.33 ± 0.15 h phase-delays compared with 0.83 ± 0.05 h for water-only controls (n = 11 per group).
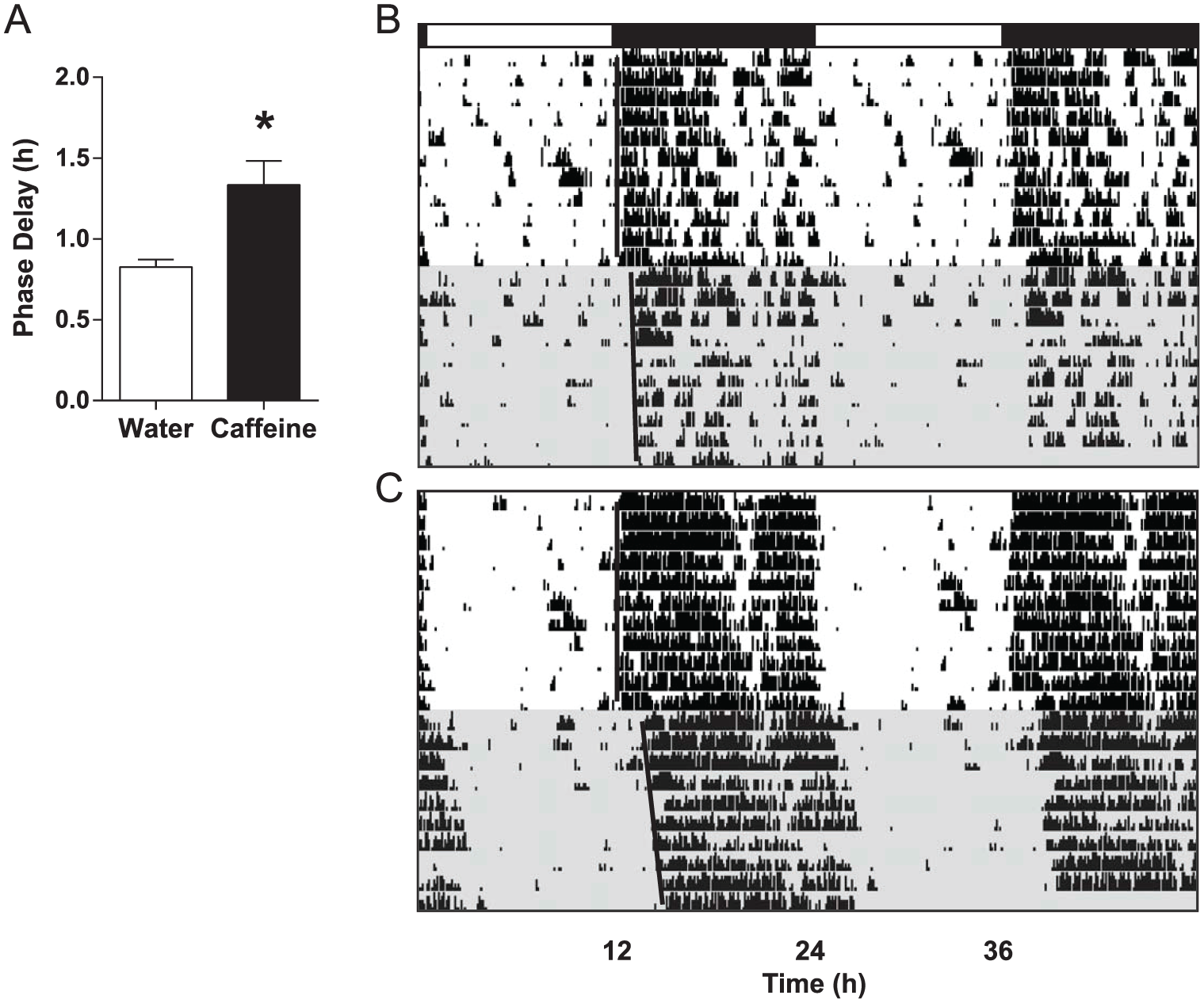
Caffeine consumption potentiates photic phase-delays in mice. (A) Compared with water-drinking controls, caffeine drinkers (with an average consumption = 103.97 ± 5.33 mg/kg/day) showed greater circadian phase-resetting responses to a 30-min light pulse delivered at ZT14. Bars show mean ± SEM, *p < 0.05. (B and C) Representative, double-plotted actograms from a water drinker and a caffeine drinker, respectively. Black bar denotes the dark phase; shaded regions represent constant darkness.
Acute Caffeine Dose-dependently Enhances Photic Phase-delays
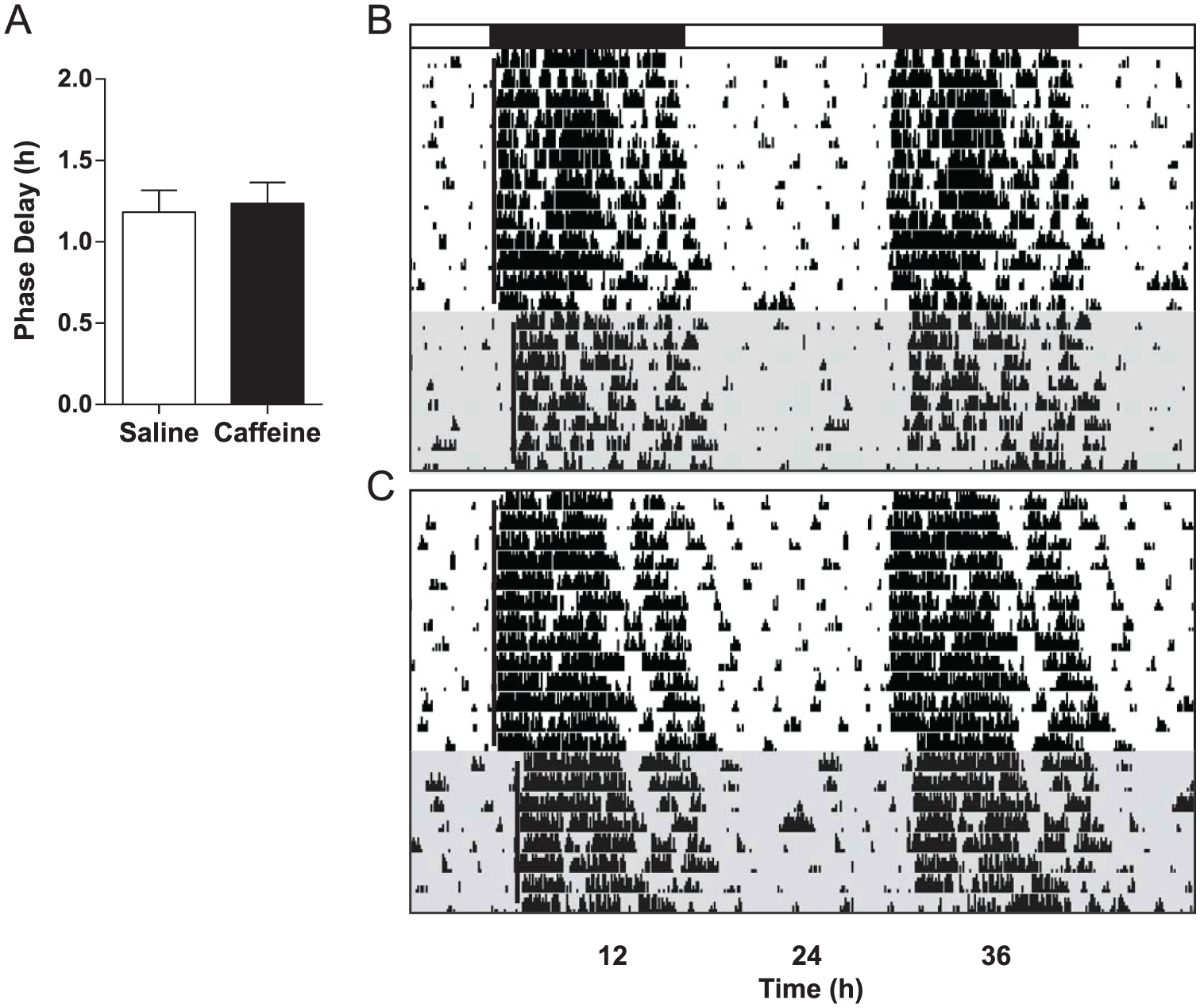
Low-dose caffeine (3.0 mg/kg) had no effect on phase-resetting in mice (n = 8) compared with saline-injected controls (n = 7) in cohort 3 (t = 0.2874, p = 0.7783) (Fig. 4), with phase-delays averaging 1.24 ± 0.13 h and 1.18 ± 0.13 h, respectively. In contrast, mice receiving high-dose caffeine (15.0 mg/kg; n = 11) showed larger phase-delay responses (0.94 ± 0.15 h) compared with saline controls (n = 9; 0.37 ± 0.10 h) in cohort 4 (t = 3.046, p = 0.007) (Fig. 5). Caffeine did not have a phase-resetting effect of its own at ZT14 in the absence of a light pulse (t = 0.2104, p = 0.8356) (Fig. 6); mice receiving caffeine (15.0 mg/kg; n = 11) but no light pulse had an average phase-shift of 0.00 ± 0.08 h, while those receiving saline (n = 10) averaged 0.02 ± 0.06 h.
Low-dose caffeine has no effect on photic phase-resetting. (A) Photic phase-delay responses were similar in mice that received either caffeine (3.0 mg/kg) or saline. Bars show mean ± SEM. (B and C) Representative, double-plotted actograms from a saline control mouse and a caffeine-injected mouse, respectively. Black bar denotes the dark phase; shaded regions represent constant darkness.
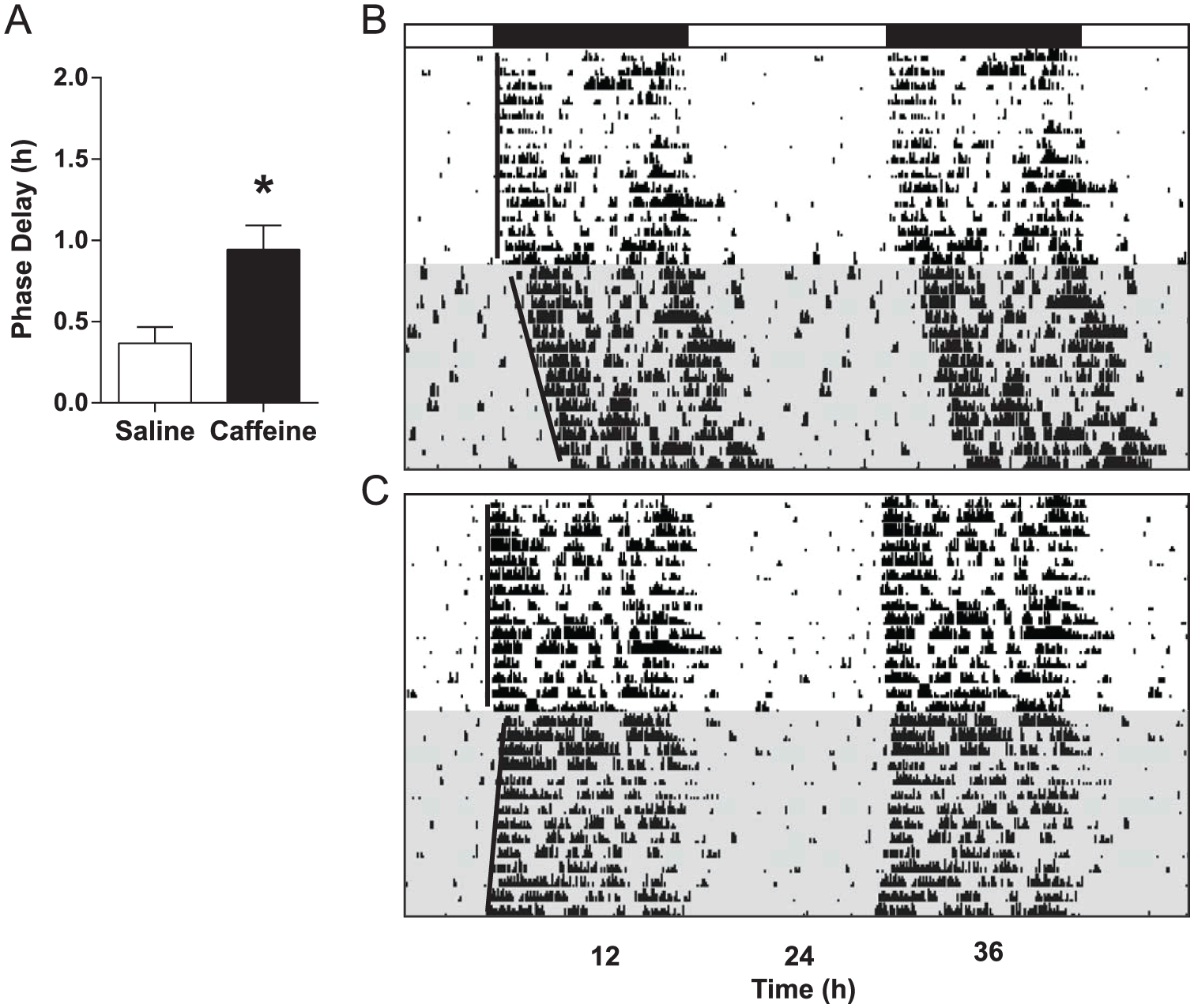
High-dose caffeine enhances phase-resetting responses to light. (A) Photic phase-delays were greater in mice that received caffeine (15.0 mg/kg) compared with those receiving saline. Bars show mean ± SEM, *p < 0.05. (B and C) Representative, double-plotted actograms from a saline control mouse and a caffeine-injected mouse, respectively. Black bar denotes the dark phase; shaded regions represent constant darkness.
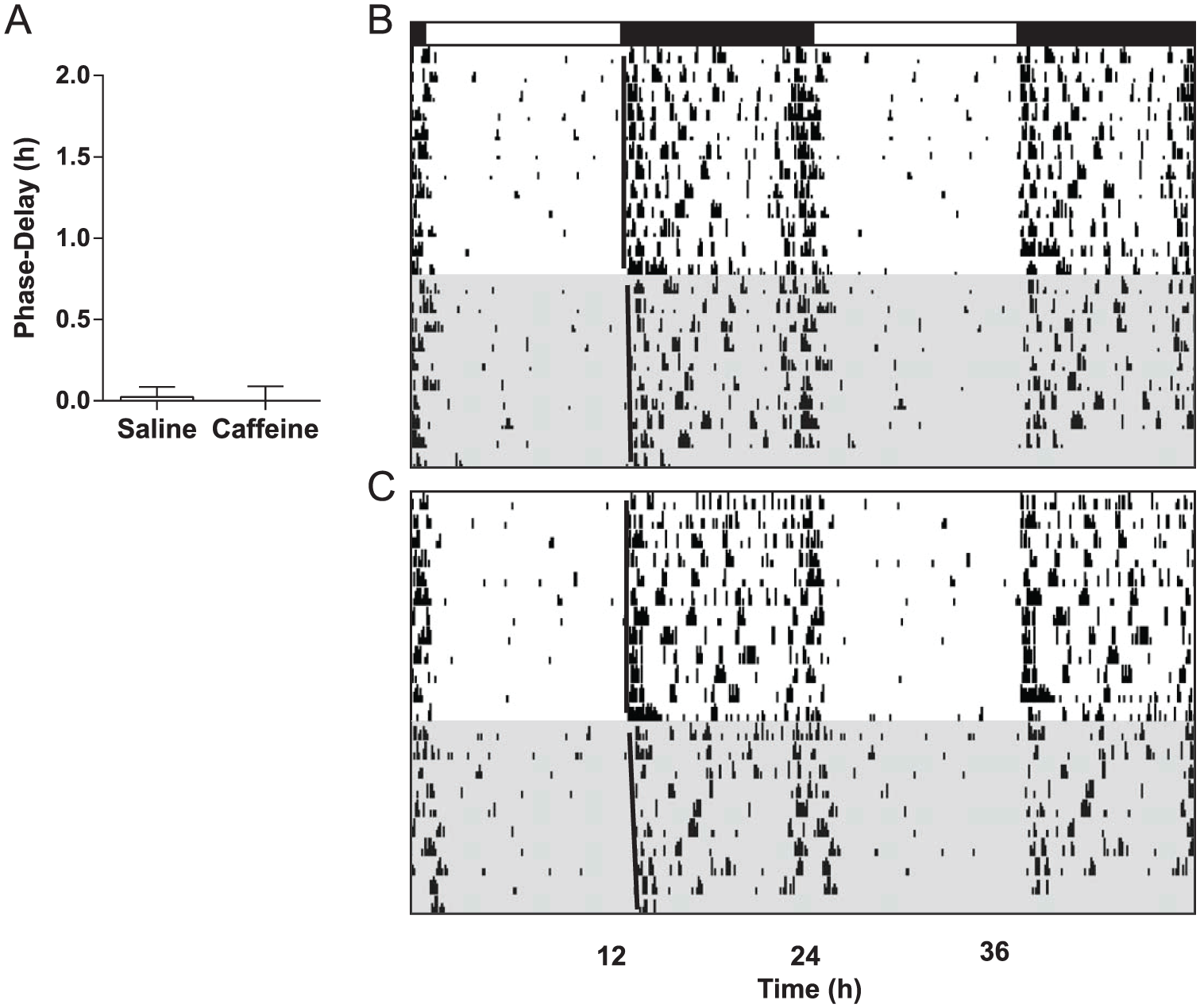
High-dose caffeine has no phase-delaying effect of its own in the early night. (A) Neither mice receiving saline nor those receiving caffeine (15.0 mg/kg) at ZT14 showed phase-resetting responses in the absence of a light pulse. (B and C) Representative, double-plotted actograms from a saline control mouse and a caffeine-injected mouse, respectively. Black bar denotes the dark phase; shaded regions represent constant darkness.
Adenosine A1, but Not A2A, Receptor Antagonism Mimics Caffeine-induced Enhancement of Photic Phase-delays
The adenosine A1 receptor antagonist DPCPX (6.0 mg/kg), but not the A2A receptor antagonist ZM-241385 (20.0 mg/kg), potentiated circadian photic phase-delays in mice compared with those measured in vehicle-treated controls (F2,19 = 6.551, p = 0.0069) (Fig. 7). Average phase-delays were 1.63 ± 0.11 h, 1.06 ± 0.10 h, and 1.20 ± 0.12 h, respectively (n = 8 per group).
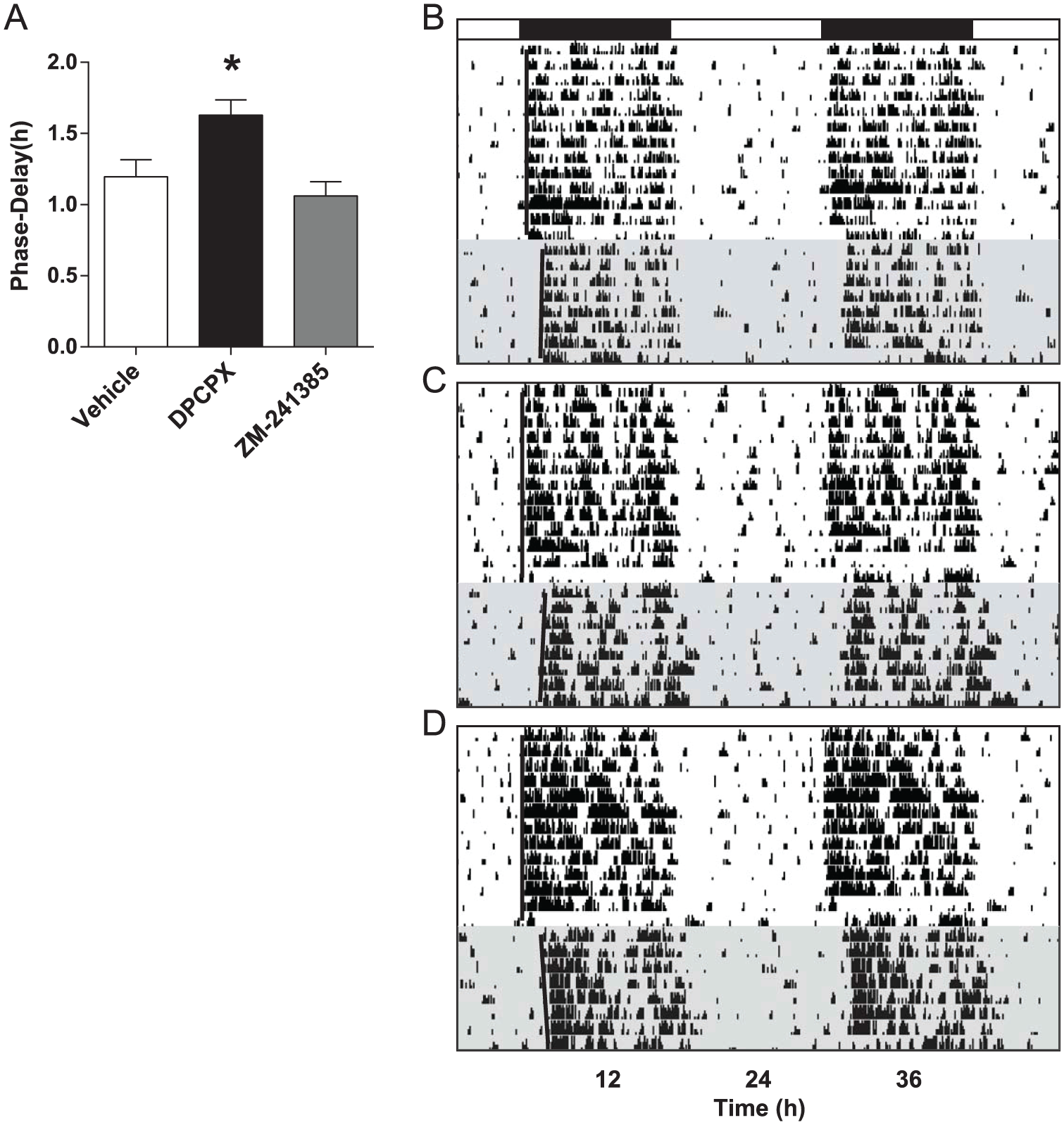
Adenosine A1 receptor antagonism mimics the effect of caffeine on photic phase-resetting. (A) Adenosine A1 receptor antagonist DPCPX (6.0 mg/kg), but not A2A receptor antagonist ZM-241385 (20.0 mg/kg), increased photic phase-delay responses in mice compared with vehicle. Bars show mean ± SEM, *p < 0.05. (B, C, D) Representative, double-plotted actograms from vehicle control, DPCPX-injected, and ZM-241385-injected mice, respectively. Black bar denotes the dark phase; shaded regions represent constant darkness.
Discussion
Here we report that caffeine consumption delays light-entrained activity in mice, and we provide evidence that it may do so by potentiating photic phase-delays. Furthermore, we demonstrate similar enhancement of photic phase-delays with a selective adenosine A1 receptor antagonist, suggesting this is the mechanism by which caffeine modulates clock timing. Our study adds to the body of evidence that caffeine not only affects the homeostatic regulation of the sleep-wake cycle but also interacts with mammalian circadian timekeeping processes to alter daily activity patterns.
Despite differences in species, experiment design, caffeine administration method, and dependent variables, our data showing delayed light entrained circadian activity in mice are remarkably consistent with a recent study by Burke et al. (2015), who showed that evening caffeine consumption delayed the nightly onset in melatonin levels in 5 human subjects. In this study, participants consumed a single dose of caffeine 3 h prior to their habitual bedtime, which resulted in an approximately 40-min delay in the melatonin rhythm. It is noteworthy that the dose of caffeine they used was somewhat comparable to the higher acute dose of caffeine we administered in the photic phase-resetting experiment. Comparing drug doses between mice and humans requires estimating metabolic rate (B) based on body mass in kilograms (M): For humans, this is best approximated by the equation B ~ M0.75 (otherwise known as Kleiber’s law), and for mice, it is best approximated by the equation B ~ M0.67 (Banavar et al., 2010; Dodds et al., 2001). Thus, the caffeine doses with behavioral effects in the two studies for a 69-kg human are approximately equal to that found in 1 to 2 brewed cups (1 cup = 8 ounces or 237 mL) of coffee (200 mg in the Burke et al. study; murine equivalent = 9 mg/kg) or 2 to 4 brewed cups of coffee (359 mg in our study). Our low dose of caffeine would be equivalent to 72 mg for a 69-kg human, roughly equivalent to that found in a strong black tea. When coffee was available in their drinking water, mice (~30 g) consumed the human equivalent of 14 to 25 brewed cups of coffee per day, which likely explains in part the larger magnitude of delay we saw herein; mice became active well over an hour later during caffeine exposure compared with their baseline. The offset of activity (e.g., onset of the rest phase) was delayed by ~40 min in our mice, which is ostensibly consistent with the well-known wake-promoting effect of caffeine and delay in sleep onset reported in other studies (Hindmarch et al., 2000; Paterson et al., 2007). However, we found a slight but significant—and very unexpected—reduction in light phase activity during caffeine exposure. Thus, although we did not measure sleep directly, we did not observe evidence that sleep is disrupted by caffeine consumption—quite the opposite. Also surprising was the lack of effect of caffeine on dark phase activity. Taken together, these data suggest an important role of caffeine in affecting circadian regulation of the sleep-wake cycle.
Our data support the hypothesis that chronic caffeine may delay light entrainment in part by enhancing photic phase-delays, an effect which appears to be due to caffeine’s antagonism of adenosine A1 receptors in the SCN (see below). As with most pharmacological agents, the pharmacodynamics of caffeine vary as a function of dose, with lower, physiologically relevant levels affecting primarily A1 and A2A receptors, while higher levels may affect additional targets (e.g., Fredholm, 1985). It is possible that mice consumed enough caffeine in that experiment to have effects not only on adenosine receptors but also on ryanodine receptors and possibly phosphodiesterase, both of which would be predicted to increase phase-delays (see below). However, it is important to consider that caffeine consumption was likely taking place primarily over their ~12-h active phase, not all at once. Previous studies examining alcohol intake in rodents have revealed a peak in consumption and resulting peripheral and SCN ethanol levels beginning around 3 h into the dark phase (Brager et al., 2010; Rhodes et al., 2005; Ruby et al., 2009). The drinking pattern for caffeine may differ from that of alcohol, and caffeine metabolism may also be subject to circadian regulation. Thus, future studies examining drinking patterns and levels of caffeine as well as its metabolites would be informative. It is also important to note that we chose to continue caffeine availability in DD to avoid inducing caffeine withdrawal, so there is a possibility that period lengthening may also contribute to the large phase-delay (Burke et al., 2015; Oike et al., 2011; van Diepen et al., 2014). Another consideration is that we used an Aschoff Type II protocol, testing mice that were in LD rather than DD prior to the light pulse. Given delayed entrainment in caffeine drinkers compared with water drinkers, the light pulse may have hit the caffeine drinkers earlier in their phase-response curve. However, we predict this would reduce, not increase, the phase-shift, and similar results with a single high dose of caffeine administered to caffeine-naïve mice support our hypothesis.
To our knowledge, we are the first to report an effect of acute caffeine at a relatively moderate dose on photic phase-resetting per se, although our results were predicted and are supported by the work of Deboer (van Diepen et al., 2014). Both experimenter-administered injections of high-dose caffeine and self-administered oral caffeine potentiated photic phase-delays in mice, suggesting this may be partly responsible for the delay in light-entrained circadian activity. Our results are in agreement with a recent study showing potentiated photic phase-resetting in the diurnal Sudanian grass rat (Jha et al., 2017), albeit with a much higher dose of caffeine (30 mg/kg; 718 mg for a 69-kg human). In contrast to our work, another group (Vivanco et al., 2013) reported an attenuating effect of caffeine on photic phase-resetting in mice, but only at a very high dose (40 mg/kg; 958 mg of caffeine for a 69-kg human). This raises the interesting possibility that caffeine may have a dose-dependent, biphasic effect on light resetting of the clock, perhaps involving multiple mechanisms, or may reflect toxicity occurring at very high doses. Caffeine is also known to affect nonphotic phase-resetting; a study by Antle et al. (2001) in Syrian hamsters showed that caffeine attenuated nonphotic phase-resetting to a 3-h sleep deprivation procedure. Thus, it is possible that enhancement of photic phase-delays and inhibition of nonphotic phase-advances could combine to delay circadian activity patterns. However, it is not likely that mice were actively consuming caffeine during the light phase (their rest phase), when sensitivity to nonphotic stimuli peaks. Thus, it is reasonable to speculate that caffeine would have more influence on photic sensitivity in mice, which occurs during the night when they were likely consuming it.
Our data also suggest that enhancement of photic sensitivity may have contributed to the melatonin phase-delay reported by Burke et al. (2015), as the human subjects in this study ingested caffeine 3 h prior to their habitual bedtime, under dim light (~1.9 lux), not darkness; participants’ history of light exposure could also have influenced light sensitivity. These authors did not observe enhanced shifting when bright light (~3000 lux) alone was compared with caffeine and bright light, but this may reflect a ceiling effect of single-pulse bright light responsiveness (Beersma and Daan, 1993; Burke et al., 2015). In the present study, we administered a relatively dim, ~25-lux light pulse. Also arguing in favor of this notion is the lack of phase-resetting effect of caffeine on its own in either the present study or others (Antle et al., 2001; Jha et al., 2017; Vivanco et al., 2013). Thus, caffeine appears to modulate responsiveness to circadian time cues rather than act as a time cue itself in vivo (although it theoretically could affect molecular clockwork in cells expressing A1 or A2A receptors). Finally, the similar action of acute and chronic caffeine on photic phase-delays indicates that tolerance to this particular effect of caffeine does not develop.
Our data showing potentiation of photic phase-delays with A1 receptor antagonist DPCPX support the hypothesis that caffeine’s action is mediated by A1 receptor inhibition. This finding is consistent with the line of evidence that A1 receptors gate retinohypothalamic photic input to the SCN (Elliott et al., 2001; Hallworth et al., 2002; Sigworth and Rea, 2003). Likewise, the lack of effect of A2A receptor antagonist ZM-241385 on photic phase-resetting is not surprising given that A2A receptors do not appear to be highly expressed in the hypothalamus (Svenningsson et al., 1999). Another mechanism by which caffeine has been shown to act is via increasing ryanodine receptor-induced mobilization of intracellular calcium (McPherson et al., 1991). Ryanodine receptors are known mediators of photic phase-delays in the SCN; caffeine induces phase-delays (CT14), but not phase-advances (CT20), in the SCN slice (Diaz-Munoz et al., 1999; Ding et al., 1998). Caffeine is also well-known for phosphodiesterase inhibition. However, supraphysiological concentrations of caffeine are required to affect ryanodine receptors and phosphodiesterases; rather, A1 and A2A receptor inhibition is responsible for caffeine’s effects at levels resulting from actual consumption (Fredholm, 1985). Without directly measuring SCN caffeine concentrations resulting from oral or intraperitoneal administration in mice, it is difficult to determine whether ryanodine receptors may play a role in our observations. Interestingly, Vivanco et al. (2013) showed the opposite effect on photic phase-delays at very high doses of caffeine (40 mg/kg), suggesting a more complex mechanism. However, in agreement with our data, Burke et al. (2015) showed that A1 receptors mediated caffeine’s delaying effect on human circadian timing, ruling out both ryanodine receptors and phosphodiesterases. Moreover, we, Antle et al. (2001), Jha et al. (2017), and Vivanco et al. (2013) showed no phase-resetting effect of caffeine on its own, arguing against these other mechanisms. Finally, Jha et al. (2017) reported caffeine-induced enhancement of photic phase-advances, which are not mediated by ryanodine receptors (Diaz-Munoz et al., 1999; Ding et al., 1998), in diurnal rodents. We are currently examining the effects of caffeine on photic phase-advances in mice, which should help settle this debate, as well as the notion that caffeine’s effects may be secondary to arousal and dependent on phase preference (Jha et al., 2017). Rather, we suggest that caffeine’s effects on circadian rhythms reflect its variable pharmacodynamic profile as a function of dose. The development of full phase-response (and dose-response) curves for caffeine, with and without light pulses, would settle this debate definitively.
We acknowledge a few limitations to the present study. First, we observed differences between saline control groups in the studies examining the impact of acute caffeine on photic phase-resetting, which is why the results of each experiment were reported separately. This is likely due to the fact that our circadian chamber holds a maximum of 24 mice, and thus some degree of variability between cohorts in individual phase-resetting responses is not unexpected. This also illustrates the importance of including vehicle controls in each phase-resetting experiment, as we did in this study (and nearly always do). Second, we did not examine the effect of caffeine on photic phase-advances in the present study due to the fact that mice show very small phase-advances to light. We are trying this experiment anyway and may in the future also include another nocturnal species with a larger phase-advance portion of the photic phase-response curve (e.g., hamsters). Third, we used only male mice in the present experiments so we cannot generalize our results to females. We plan to include females in future studies as well; photic phase-resetting data from other experiments in our laboratory suggest that responses in female mice are comparable to males in both magnitude and variability. Fourth, it would have strengthened our contention that A1 receptor inhibition mediates caffeine-induced potentiation of photic phase-delays if we could have blocked caffeine’s effect by pretreating with a selective A1 receptor agonist. We performed such an experiment using selective adenosine A1 and A2A receptor agonists, but the sedation produced by the A1 receptor agonist confounded interpretation of the results and thus we did not report them. We may revisit this question in the future using another method.
In summary, we show that delayed light-entrained circadian activity rhythms during caffeine consumption may be due to potentiation of photic phase-delays mediated by A1 receptor inhibition. We also note the absence of evidence for increased rest-phase activity during caffeine intake in the present study. Taken together, our data suggest an important role of caffeine in affecting circadian regulation of the sleep-wake cycle. In human terms, delays in circadian activity patterns are generally unwelcome because they exacerbate the frequent sleep deprivation we too often experience in order to meet the demands of professional, social, and school schedules. However, it is important to note that health benefits are attributed to caffeine as well: for example, its negative correlation with depression risk in women (Lucas et al., 2011) and prevention of cognitive decline in the elderly (Panza et al., 2015). Thus, we also suggest that caffeine consumption at other times of day (e.g., the morning, rather than the night) may not be harmful and may even be beneficial. We plan to test this hypothesis in future studies.
Footnotes
Acknowledgements
This research was supported by the College of Natural Sciences and Mathematics and the Department of Biology at Indiana University of Pennsylvania. The authors declare no competing financial interests.
Conflict of Interest Statement
The authors have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.