
Abstract
In nocturnal rodents, voluntary wheel-running activity (WRA) represents a self-reinforcing behavior. We have previously demonstrated that WRA is markedly reduced in mice with a region-specific deletion of the transcription factor Pou4f1 (Brn3a), which leads to an ablation of the dorsal medial habenula (dMHb). The decrease in WRA in these dMHb-lesioned (dMHbCKO) mice suggests that the dMHb constitutes a critical center for conveying reinforcement by exercise. However, WRA also represents a prominent output of the circadian system, and the possibility remains that the dMHb is a source of input to the master circadian pacemaker located in the suprachiasmatic nucleus (SCN) of the hypothalamus. To test this hypothesis, we assessed the integrity of the circadian system in dMHbCKO mice. Here we show that the developmental lesion of the dMHb reduces WRA under both a light-dark cycle and constant darkness, increases the circadian period of WRA, but has no effect on the circadian amplitude or period of home cage activity or the daily amplitude of sleep stages, suggesting that the lengthening of period is a result of the decreased WRA in the mutant mice. Polysomnographic sleep recordings show that dMHbCKO mice have an overall unaltered daily amplitude of sleep stages but have fragmented sleep and an overall increase in total rapid eye movement (REM) sleep. Photoresponsiveness is intact in dMHbCKO mice, but compared with control animals, they reentrain faster to a 6-h abrupt phase delay protocol. Circadian changes in WRA of dMHbCKO mice do not appear to emerge within the central pacemaker, as circadian expression of the clock genes Per1 and Per2 within the SCN is normal. We do find some evidence for fragmented sleep and an overall increase in total REM sleep, supporting a model in which the dMHb is part of the neural circuitry encoding motivation and involved in the manifestation of some of the symptoms of depression.
Circadian pacemakers have an endogenous period close to 24 h in the absence of environmental cues and can be reset by entraining to daily environmental cycles. In mammals, behaviors such as wheel-running activity (WRA) often provide temporal feedback to the circadian system to reset the clock or regulate the period of the pacemaker (Edgar and Dement, 1991; Edgar et al., 1991; Mistlberger, 1991; Yamada et al., 1988). We have recently shown that the dorsal medial habenula (dMHb) is essential for normal levels of voluntary WRA and that reduced WRA in dMHb-lesioned (dMHbCKO) mice may be accounted for by a lack of perceived reinforcement (Hsu et al., 2014). However, a reduction in the amplitude of circadian WRA can result from decreased in vivo suprachiasmatic nucleus (SCN) electrical activity and clock gene expression in mice (Han et al., 2012; Nakamura et al., 2011). Thus, the reduced WRA seen in dMHbCKO mice could also be a result of reduced activity feedback to the circadian system and/or decreased amplitude in the circadian circuitry involving the SCN and the habenula, and not solely due to reduced motivation.
The habenula is located on the dorsal surface of the thalamus; this paired structure has been implicated in the regulation of a wide variety of behaviors, including motivation, reward, learning and memory, circadian rhythms, and sleep (Aizawa et al., 2013; Proulx et al., 2014; Valjakka et al., 1998; Velasquez et al., 2014; Zhao et al., 2015). The habenula neurons project to the interpeduncular nucleus through the fasciculus retroflexus (FR). Both transection of the FR in hamsters (Paul et al., 2011) as well as dMHb ablation in mice (Hsu et al., 2014) reduce the amount of WRA and change its 24-h temporal distribution. In contrast, there is no change in locomotion in an open field or in the home cage between control and dMHbCKO mice (Hsu et al., 2014). Interestingly, the circadian periods of both WRA and home cage activity (HCA) were longer in FR lesioned hamsters compared with control animals (Paul et al., 2011). These findings, together with the fact that the habenula exhibits autonomous rhythms of gene expression and electrical activity (Guilding et al., 2010; Sakhi et al., 2014; Tavakoli-Nezhad and Schwartz, 2006; Zhao and Rusak, 2005), have suggested that the habenula contains an extra-SCN circadian oscillator involved in the regulation of circadian rest-activity cycles. However, the functions of the main subregions of the habenula as part of the circadian system remain to be determined.
In the present study, we addressed these questions by first measuring voluntary WRA and HCA of intact and dMHb-lesioned mice under both 12:12 LD cycle and constant darkness (DD) and determining whether the lesion affects circadian period. We also examined photoresponsiveness by looking at light-induced phase shifting of WRA and reentrainment after abrupt LD cycle phase shifts. Finally, we measured the circadian expression of SCN Per1 and Per2, as well as electroencephalographic (EEG)–recorded sleep stages to determine if the central pacemaker and another of its prominent circadian outputs were affected by the lesion. Together, our results show that genetic ablation of the dMHb has minimal impact on the circadian regulation of locomotor activity and sleep. Although the circadian period of WRA is lengthened in dMHbCKO mice, this change appears to be a consequence of the decrease in WRA that may result from reduced motivation. On the other hand, ablation of the dMHb leads to increases in sleep fragmentation and in rapid eye movement (REM). These sleep abnormalities, together with reduced perceived reinforcement of locomotor activity and a range of both normal and abnormal depression-related behavioral tests we have previously described (Hsu et al., 2016; Hsu et al., 2014), suggest that the loss of dMHb function leads to a specific subset of the canonical manifestations of depression.
Materials and Methods
Generation of Transgenic Mice
All experiments with animals were performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee of the University of Washington and the Seattle Children’s Research Institute.
Mice with a tissue-specific null mutation of Pou4f1 in the dMHb and control littermates used in this study were generated and genotyped as previously described (Hsu et al., 2014). Three transgenic strains of mice were used: Pou4f1tlacZ, in which a functionally null allele replacing the Pou4f1 coding sequence with a β-galactosidase expression cassette (Quina et al., 2005); Pou4f1flox, in which the principal coding exon of Pou4f1 is flanked by loxP sequences (Gerfen et al., 2013; Hsu et al., 2014); and Syt6Cre, STOCK Tg(Syt6-Cre)KI148Gsat/Mmcd, a BAC transgenic line obtained from the Gensat project. Experimental mice were generated by crossing mice with the genotype Pou4f1tlacZ/+, Syt6Cre/Cre with Pou4f1flox/flox mice to yield the genotypes Pou4f1tlacZ/flox, Syt6Cre/+ (dMHbCKO) and Pou4f1flox/+, Syt6Cre/+ (control) mice in equal ratios. The dMHbCKO mice show a profound loss of neurons in the dMHb due to postnatal cell death, while control mice do not show detectable loss of dMHb neurons (Hsu et al., 2014). Pou4f1+/− and Pou4f1+/+ mice were generated by crossing mice with the genotype Pou4f1tlacZ/+, Syt6Cre/Cre with C57BL/6NCrl mice (Charles River Laboratories). All mice used were backcrossed >6 generations to C57BL/6NCrl mice. Behavioral assays were performed on male mice 2 to 8 mo of age.
Behavioral Measurements and Analyses
For WRA, mice were individually housed in cages equipped with a horizontal wire-running wheel and maintained in a ventilated, light-tight room under a 12:12 LD cycle with 200-lux intensity or DD. WRA was monitored using Clocklab software (Actimetrics) and analyzed using El Temps software (Dr. Toni Diéz-Noguera, Universidad de Barcelona) as previously described (Han et al., 2012). Data were analyzed in 10-min or 60-min bins for double-plotted WRA actograms or WRA waveforms, respectively.
To determine if light would induce phase shift of circadian WRA, lights were turned off following assessment of LD WRA, and mice remained in DD throughout a 24-h period. Mice remained in their home cages and were moved to another chamber, exposed to a 30-min pulse of light (500-lux) at circadian time (CT) 16 (with CT12 defined as the onset of WRA and each circadian hour approximated to 60 min), and transferred back to the chamber in DD. To quantify the light-induced phase delay, a line through the onset of WRA was drawn on the plotted actograms by 3 independent observers for the 6 to 8 cycles after exposure to the light pulse and then extrapolated back to the day of the pulse; the delay was measured as the difference between the activity onset on the day of the pulse and the extrapolated line.
To determine the effect of abrupt LD phase shifts on the phase of the circadian WRA rhythm, a delay and advance “jetlag” protocol was employed (Han et al., 2012). After mice were entrained to a 12:12 LD cycle, a delay jetlag was initiated by extending the light phase by 6 h (i.e., delaying the time of lights-off by 6 h). The mice stayed under this new 12:12 LD cycle until they were reentrained. The advance jetlag was initiated by shortening the light phase by 6 h (i.e., advancing the time of lights off by 6 h). For delays, we counted the number of days for the activity offset to reach the same phase relationship—relative to the LD cycle—as before the LD shift. For advances, we counted the number of days for activity onset to reach a same phase relationship as before the LD shift. Three independent observers assessed both delay and advance shifts, and the average day count was used.
To determine the phase angle of entrainment, the time of onset of WRA after mice were released into DD was subtracted from the time of lights-off before the release. A positive phase angle of entrainment implies the mouse started WRA prior to time of lights-off.
HCA was measured by housing mice singly in cages equipped with an infrared (IR) beam detector (Gardtec Gardscan MX PIR, RS Components Ltd., Northamptonshire, UK) and without a running wheel.
A single cohort of control and dMHbCKO mice was used for all circadian behavioral rhythms measured using WRA. The order of the tests conducted was as follows: LD WRA measurements, release into DD and CT16 light-induced phase shift, DD WRA and period measurements, reentrainment to the LD cycle, and delay jetlag followed by advance jetlag. Following administration of the CT16 light pulse, mice remained in DD until WRA had stabilized a stable period before assessments of DD WRA free-running period began (Fig. 1A, arrowhead). A second cohort of control and dMHbCKO mice was used to examine the phase angle of entrainment; both genotypes of this second cohort had undergone a sucrose preference test, a rotarod test, a Catwalk test, and an open field test reported elsewhere (Hsu et al., 2014). A third cohort was used to measure HCA. A fourth cohort of Pou4f1+/− and Pou4f1+/+ mice was used to assess the effect of Pou4f1 haploinsufficiency on WRA.
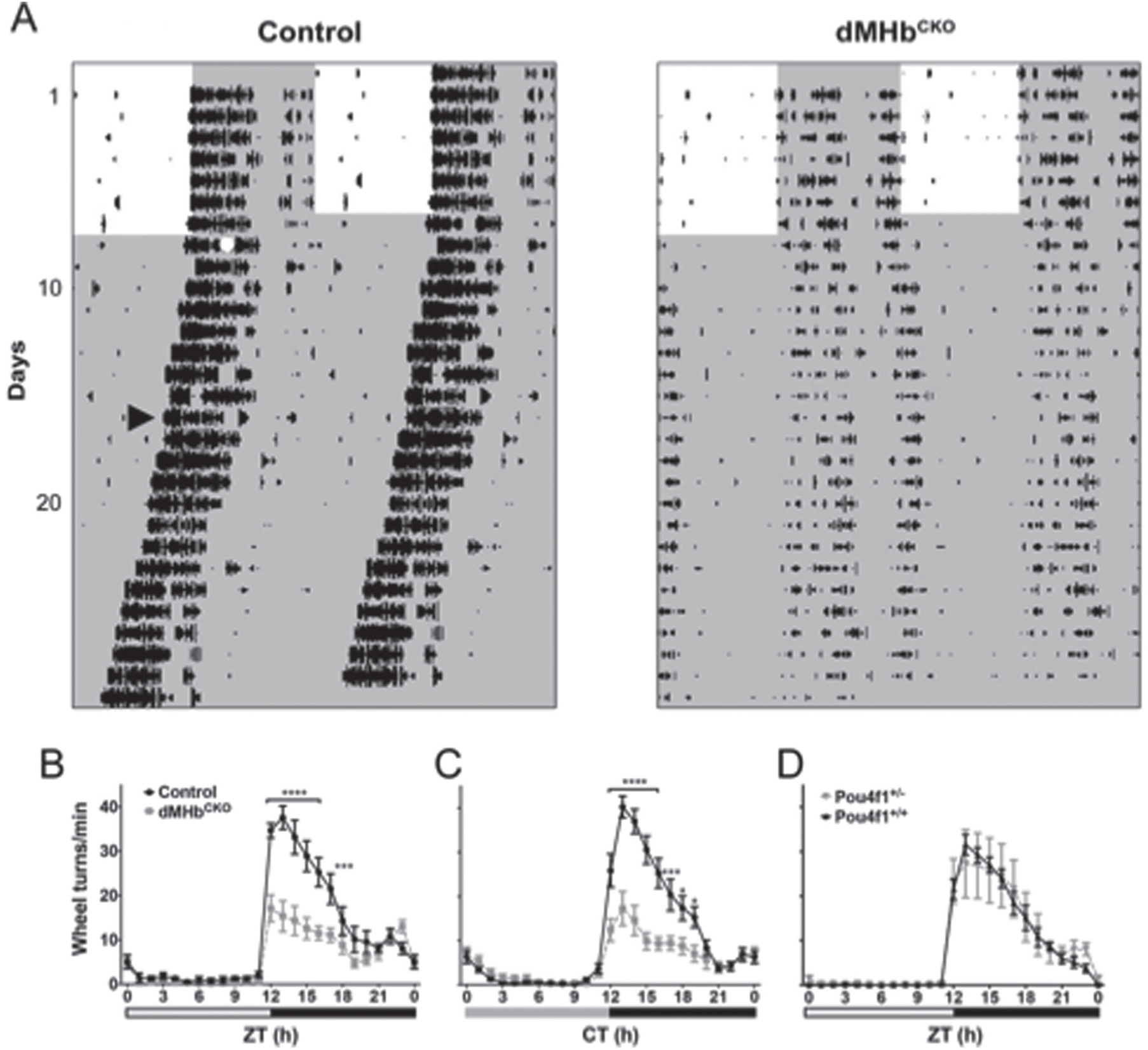
Wheel-running activity (WRA) in dMHbCKO mice. (A) WRA in representative control and dMHbCKO mice measured under a 12:12 LD cycle and DD. WRA is displayed as double-plotted actograms with wheel revolutions represented as black bars on successive days (vertical axis) plotted against the 48-h period (horizontal axis). White and gray areas represent light and dark phases, respectively. The white circle denotes a 30-min, 500-lux light pulse at circadian time 16 (CT16); the arrowhead indicates the first cycle included in the calculation of the free-running period and amplitude, after the WRA had stabilized following the CT16 light pulse. (B) Summary of WRA for control and dMHbCKO mice over a 24-h LD cycle. dMHbCKO mice showed significantly less WRA, especially during the first 6 h of the night. n = 13 control and 14 dMHbCKO mice. Six days of WRA were averaged for each of the control and dMHbCKO mice. Zeitgeber time 12 (ZT12) corresponds to time of lights-off for a 12:12 LD cycle, and white and black bars below represent light and dark phases, respectively. (C) Summary of WRA for control and dMHbCKO mice under DD. n = 11 control and 12 dMHbCKO mice. Ten to 14 d of WRA were averaged for each of the control and dMHbCKO mice, using each animal’s period as the 24-h circadian period. CT12 corresponds to time of activity onset; gray and black bars below represent subjective day and night, respectively. (D) WRA is not affected in Pou4f1+/− mice. Summary of WRA for Pou4f1+/− and Pou4f1+/+ mice over a 24-h LD cycle. Both genotypes showed a similar amount of WRA. n = 10 Pou4f1+/+ and 5 Pou4f1+/− mice. Ten to 12 d of WRA were averaged for each of the Pou4f1+/+ and Pou4f1+/− mice, with the exception of 1 Pou4f1+/+ mouse for which 5 d of WRA were averaged. Symbols as in (B). Values for ZT0 (B, D) and CT0 (C) are replotted at the end for easier visualization. ****p < 0.0001, ***p < 0.001, *p < 0.05, significant difference between genotypes.
Sleep
Sleep stages were recorded as previously described with some modifications (Cambras et al., 2007; Lee et al., 2009). Mice were anesthetized with isoflurane and placed into a stereotaxic device where isoflurane anesthesia continued throughout the surgery protocol. Each mouse was implanted with EEG electrodes, consisting of dental screws (Pinnacle Technology, Lawrence, KS; No. 8209: 0.10-in.). A midline incision was made above the skull. Recording electrodes were screwed through cranial holes as follows: over the left frontal cortex (1.5 mm lateral and 2 mm anterior to bregma) and over the right parietal cortex (1.5 mm lateral and 2 mm posterior to bregma), a ground electrode was placed over the visual cortex (1.5 mm lateral and 4.0 mm posterior to bregma), and a reference electrode was placed over the cerebellum (1.5 mm lateral and 6.5 mm posterior to bregma). Electromyogram (EMG) signals were obtained by placing a pair of silver wires into the neck muscles. The screws were connected, through silver wires, to a common 6-pin connector compatible with our recording device (Pinnacle Technology). The screws and connector were fixed to the skull with dental cement. After surgery, mice were housed in single recording cages under a 12:12 LD cycle.
After a recovery period of 5 to 7 d, mice were connected to the data-recording device. To allow mice to acclimate to the preamplifier and tether, we waited another 2 d after connecting them to start recording data. Two days of EEG and EMG activity were recorded under a 12:12 LD cycle. The EEG and EMG signals were sampled at 400 Hz with an EEG low-pass filter of 25 Hz and a low-pass EMG filter of 100 Hz. Each 10-s epoch was classified as either wake, non–rapid eye movement (NREM) sleep, or REM sleep using an automatic scoring algorithm written in R version 3.3.0. First, the power spectrum of the EEG and EMG signals is calculated using an overlap window of 50%. Then, for each 10-s epoch, the following parameters are generated: (1) delta power: the sum of EEG power at frequencies ranging between 0.5 and 4 Hz; (2) theta power: the sum of EEG power at frequencies ranging between 6 and 12 Hz; and (3) EMG RMS: the mean square root of the raw EMG signal. For each 10-s epoch, we determined the brain state using the following algorithm: if EMG RMS was higher than a threshold visually determined for each 24-h recording session, the epoch was classified as wake; otherwise, the epoch was defined as sleep. Then, sleep stage was classified as REM whenever the theta to delta power ratio during the epoch was higher than the average theta to delta ratio—over the whole 24-h recording session—plus 1 SD. The remaining epochs were classified as NREM sleep. This scoring method was based on previous studies (Fenzl et al., 2007; Louis et al., 2004; Weber et al., 2015), executed through a newly developed R code (Ben-Hamo et al., unpublished) and validated by 2 independent experimenters that visually scored 10-s bins as previously described (Cambras et al., 2007; Lee et al., 2016; Lee et al., 2009), with a minimum of 90% score agreement in both cases. The 10-s scored epochs were further binned into 20- or 60-min bins, for which we calculated the percentage of each brain state. A 20- or 60-min bin was classified as REM when more than 20% of the bin was spent in REM sleep. If REM sleep was lower than 20% during the bin, it was classified as either wake or NREM sleep, depending on which of the 2 brain states contributed to a higher percentage of the 20- or 60-min bin. Hypnograms were generated using the 10-min bin sleep scores to feed a custom-made “R” code (Ben-Hamo et al., unpublished). REM intensity was calculated as the product of REM percentage and theta power within each 20- or 60-min bin. Statistical analysis was performed for both 20- and 60-min bins and yielded similar results; only 60-min bin results are reported.
In Situ Hybridization
Mice were housed in cages provided with a running wheel under DD conditions and sacrificed every 4 h over a 24-h period at the indicated circadian times. In situ hybridization for mPer1 and mPer2 was performed with radiolabeled riboprobes as previously described (de la Iglesia, 2007). Autoradiographic images were generated by exposing hybridized brain slices to Ultramax film (Kodak). Images were scanned at high resolution, and optical densities (ODs) of the autoradiographic hybridization signals were measured with ImageJ software (National Institutes of Health) using a template for the whole SCN.
General Statistical Methods
Statistical analyses were conducted using unpaired or paired 2-tailed t tests, 2-way analysis of variance (ANOVA), or a mixed-model 2-way ANOVA, in which 1 factor (time) was used as a repeated measure. Post hoc comparisons were done with Sidak’s post hoc analyses when appropriate. To measure sleep-stage fragmentation, we used survival analysis (Lee et al., 2016; Norman et al., 2006). For each individual, we identified all periods consisting of contiguous sleep stages (total sleep, REM, or NREM sleep) in the 10-s epoch sleep scores. Following the identification of the contiguous sleep epochs for each individual, we fitted a survival curve for all control and dMHbCKO mice and used Kaplan-Meier estimates of survival to compare the 2 groups. The 24-h distribution of REM intensity was compared between phenotypes by a 2-sample Kolmogorov-Smirnov test, as the data were not normally distributed. All tests were done with GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA) or R version 3.1.2.
Results
dMHbCKO Mice Have Reduced Voluntary Wheel Running and Longer Free-Running Period
Our previous study has shown that mice with lesions of the dMHb exhibit a reduction in voluntary WRA while entrained to a 12:12 LD cycle (Hsu et al., 2014). To assess the possibility that the reduced WRA may be a result of changes in its circadian regulation, we measured WRA in control and dMHbCKO mice while they were free running under constant darkness (Fig. 1). Both genotypes entrained to the 12:12 LD cycle and dMHbCKO mice exhibited less WRA compared with control mice (interaction: F(23,575) = 10.96, p < 0.0001; genotype: F(1,25) = 15.73, p = 0.0005; time: F(23,575) = 65.66, p < 0.0001; Fig. 1B), especially during the first half of the night between zeitgeber time (ZT) 12 and ZT17 (with ZT12 the time of lights-off). In DD, this phenotype persisted in dMHbCKO mice (interaction: F(23,483) = 14.42, p < 0.0001; genotype: F(1,21) = 15.38, p = 0.0008; time: F(23,483) = 63.83, p < 0.0001; Fig. 1C), and, as under LD conditions, post hoc analyses indicated that the dMHbCKO mice exhibited less WRA during the early subjective night, between CT12 and CT19. In addition to the reduced WRA, dMHbCKO mice free run with a longer period compared with control mice (see below for 2-way ANOVA results).
Because of the nature of the genetic model used, the dMHbCKO mice used in this experiment have both copies of the Pou4f1 gene excised in the dMHb but are hemizygous for Pou4f1 elsewhere, bearing 1 copy of the constitutive null Pou4f1tlacZ allele and of the Pou4f1flox allele. To test whether the reduced WRA could be due to haploinsufficiency of Pou4f1, rather than the dMHb lesion observed in dMHbCKO mice, we also examined voluntary WRA on a cohort of matched Pou4f1+/− and Pou4f1+/+ mice. No difference in WRA was observed between Pou4f1+/− and Pou4f1+/+ mice (interaction: F(23,299) = 0.27, p = 0.99; genotype: F(1,13) = 0.23, p = 0.64; time: F(23,299) = 47.92, p < 0.0001; Fig. 1D). Thus, we conclude that the developmental loss of the dMHb, and not reduced expression of Pou4f1 elsewhere in the brain, mediates the reduced WRA observed in dMHbCKO mice.
Ablation of dMHb Neurons Affects HCA Minimally
For this experiment, we measured HCA as interruptions of an IR beam in the home cage (Fig. 2A). Confirming our previous finding (Hsu et al., 2014), there was no difference in IR crossings between control and dMHbCKO mice entrained to a 12:12 LD cycle, although the 24-h temporal distribution of HCA differed between genotypes (interaction: F(23,414) = 1.57, p = 0.047; genotype: F(1,18) = 2.81, p = 0.11; time: F(23,414) = 66.14, p < 0.0001; Fig. 2B). There was also no difference in HCA between genotypes under DD (interaction: F(23,391) = 1.04, p = 0.41; genotype: F(1,17) = 0.93, p = 0.35; time: F(23,391) = 30.79, p < 0.0001; Fig. 2C).
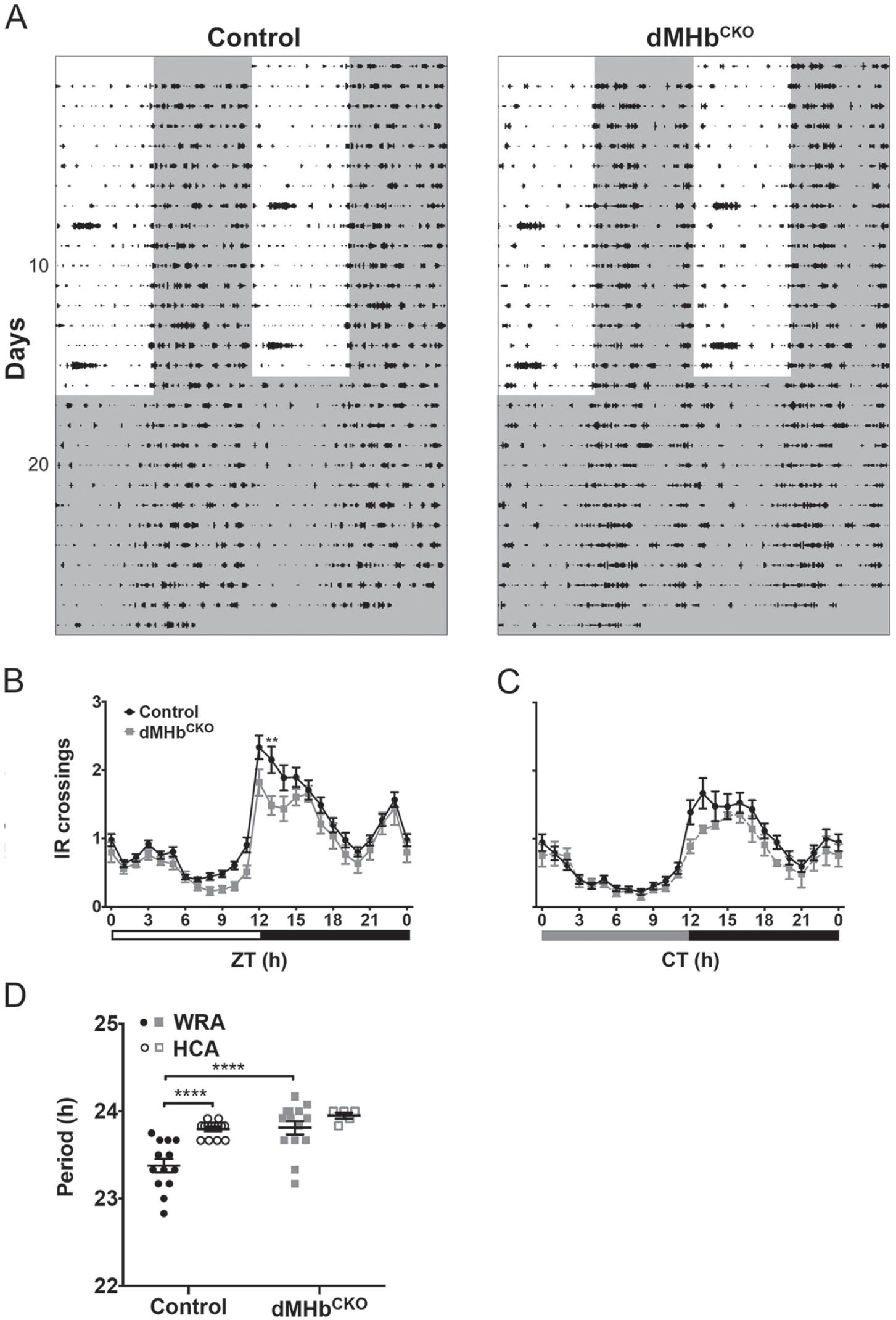
dMHbCKO mice exhibit normal general locomotion with longer circadian period. (A) General locomotor activity (home cage activity [HCA]) was measured under a 12:12 LD cycle and DD by infrared (IR) beam interruptions. HCA is displayed as double-plotted actograms with IR interruptions represented as black bars on vertical axis plotted against the 48-h period. (B) Summary of HCA for control and dMHbCKO mice over a 24-h LD cycle. Both genotypes showed similar amount of HCA, although there was a significant interaction effect (see text for details). n = 16 control and 6 dMHbCKO mice. Sixteen days of IR beam crossings were averaged for each of the control and dMHbCKO mice, with the exception of 1 dMHbCKO mouse, where 14 d of averaged IR bream crossings were used. Increased activity seen during the lights-on period on day 8 and 15 in (A) were a result of cage changing. (C) Summary of HCA for control and dMHbCKO mice during constant darkness. Both genotypes showed a similar amount of general locomotion activity. n = 11 control and 12 dMHbCKO mice. Twelve days of IR beam crossings were averaged for each of the mice. (D) dMHbCKO mice had a longer period (23.81 ± 0.076 h) than control mice (23.38 ± 0.078 h) when measured using wheel-running activity (WRA). Two separate cohorts of mice were used for the WRA (from Fig. 1) and HCA period measurements. Values for ZT0 (B) and CT0 (C) are replotted at the end for easier visualization. **p < 0.01, significant difference between genotypes. ****p < 0.0001, significant difference between genotypes/methods.
We compared the WRA and HCA period of control and dMHbCKO mice (Fig. 2D). There was an effect of genotype (F(1,42) = 16.01, p = 0.0003) and method of recording activity (F(1,42) = 14.61, p = 0.0004) but no effect of the interaction (F(1,42) = 3.67, p = 0.062). Post hoc comparisons indicated that circadian periods were different between control and dMHbCKO for WRA but not for HCA, suggesting that the effect of genotype emerged from WRA period differences.
dMHbCKO Mice Have Normal Amounts of Total Sleep but Fragmented Sleep and Increased REM Sleep
To determine if lesions of the dMHb affect outputs other than WRA, and given the proposed role of the habenula on the regulation of sleep, we were able to examine the sleep patterns in a limited number of control and dMHbCKO mice under a 12:12 LD cycle (Suppl. Fig. S1). Throughout the 24-h LD cycle (Suppl. Fig. S1A), the time spent in wake or non-REM sleep did not differ between genotypes (wake: t(5) = 0.69, p = 0.52; non-REM sleep: t(5) = 0.46, p = 0.67). In contrast, total REM sleep was higher in dMHbCKO mice than in controls (t(5) = 2.68, p = 0.0436; Suppl. Fig. S1A). We also looked at wake and sleep duration during either the light or the dark phase (Suppl. Fig. S1B). As expected, both genotypes displayed more wakefulness during the dark phase (interaction: F(1,5) = 0.04, p = 0.84; genotype: F(1,5) = 0.48, p = 0.52; time: F(1,5) = 33.84, p = 0.0021) and more non-REM sleep during the light phase (interaction: F(1,5) = 0.02, p = 0.89; genotype: F(1,5) = 0.21, p = 0.67; time: F(1,5) = 35.46, p = 0.0019; Suppl. Fig. S1B). While both genotypes displayed more REM sleep during the light phase, there was a significant effect of genotype (interaction: F(1,5) = 0.29, p = 0.61; genotype: F(1,5) = 7.21, p = 0.0436; time: F(1,5) = 23.11, p = 0.0049; Fig. S1), with dMHbCKO mice showing higher levels of REM sleep.
Although total sleep throughout the 24-h LD cycle did not differ between genotypes, the possibility exists that sleep fragmentation is affected by ablation of the dMHb. Indeed, inspection of 10-min–binned hypnograms from individuals from each genotype suggests that sleep is more fragmented in dMHbCKO mice (Suppl. Fig. S1C). We performed survival analysis and confirmed that total-sleep bouts have shorter duration in dMHbCKO mice (Suppl. Fig. S1D). Interestingly, survival analysis of non-REM and REM sleep indicated that while non-REM bouts were shorter in dMHbCKO than control mice, an opposite trend, although not statistically significant (p = 0.091), was observed for REM sleep (Suppl. Fig. S1D). In fact, this effect was statistically significant for both sleep stages in the first 24 h of sleep recordings (p < 0.001 and p = 0.03 for NREM and REM sleep, respectively). Thus, the overall increase in REM sleep found in dMHbCKO mice relative to controls is likely accounted for by an increase in REM bouts and a longer duration of these bouts. Finally, the distribution of REM intensity over the 24-h LD cycle did not differ between the 2 genotypes (Kolmogorov-Smirnov test, p = 0.063; Suppl. Fig. S1E).
dMHbCKO Mice Have Intact Circadian Photoresponsiveness
We examined photoresponsiveness first by looking at the response of WRA to a phase-shifting light pulse. A brief light pulse administered during the early subjective night is known to cause a delay in the circadian pacemaker (Johnson et al., 2003). Both genotypes were exposed to a 30-min, 500-lux light pulse at CT16, 4 h after the activity onset after 24 h in darkness (Fig. 1A, white circle). The mean phase delay for WRA onset was not significantly different for the control and dMHbCKO mice (116.6 ± 16.34 vs. 105.1 ± 12.39 min, respectively; t(25) = 0.56, p = 0.58). We also examined reentrainment to abrupt LD cycle phase shifts (Fig. 3A). dMHbCKO mice reentrained faster to a 6-h phase delay compared with control mice (t(25) = 2.36, p = 0.027; Fig. 3B), but in response to a 6-h phase advance, both genotypes were fully reentrained after a similar number of days (t(25) = 0.45, p = 0.66; Fig. 3C). Finally, the phase angle of entrainment was more variable in dMHbCKO mice (F tests for variances; F(27,22) = 4.67, p = 0.0005) but did not differ between genotypes (Mann-Whitney test, p = 0.1469).
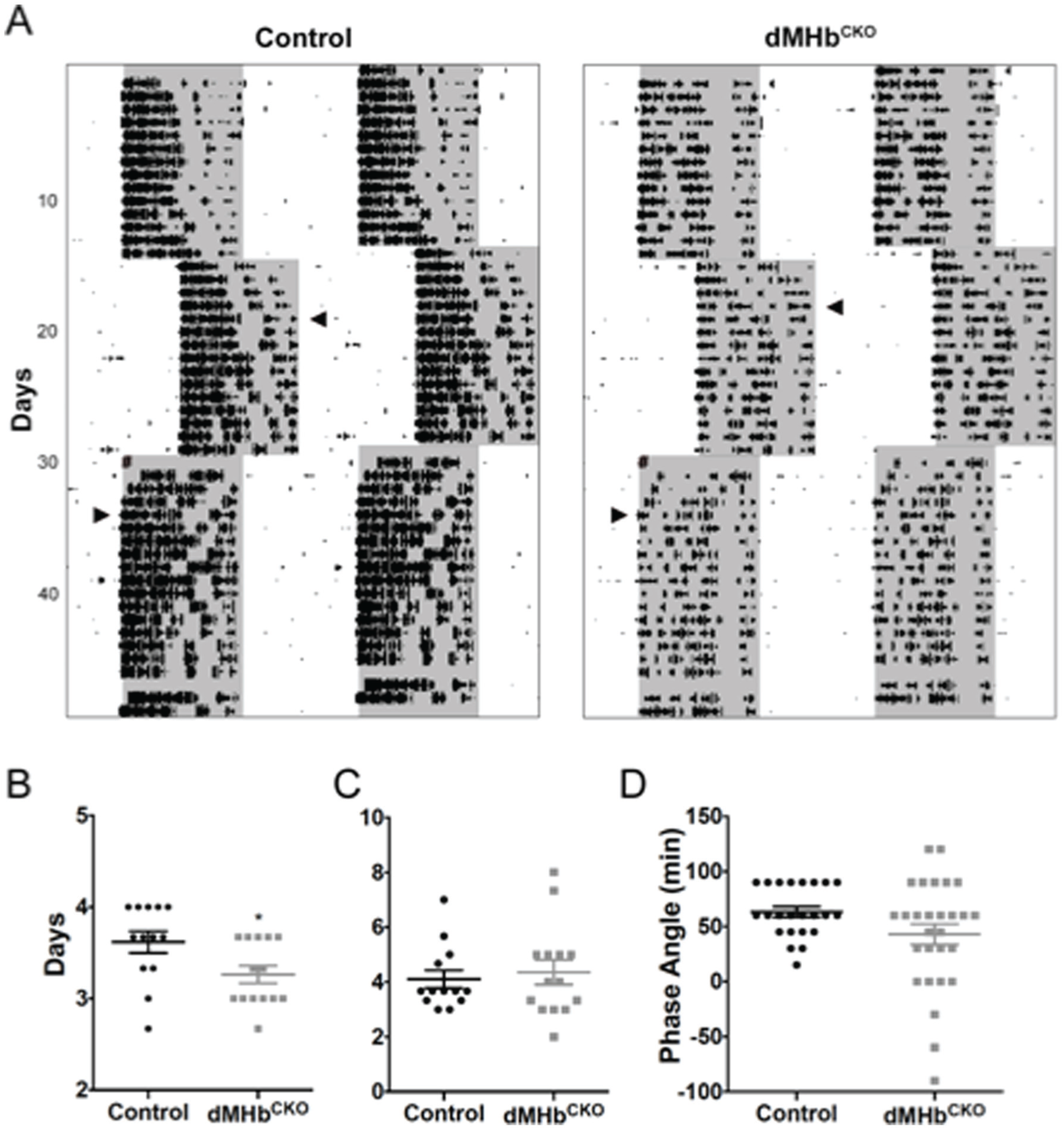
dMHbCKO mice reentrained faster to a 6-h jetlag delay of the LD cycle. (A) Representative actograms of control and dMHbCKO mice exposed to an abrupt 6-h delay and later to an abrupt 6-h advance. Wheel-running activity is plotted as described previously. Arrowheads indicate the day when the animals were fully reentrained to the new LD cycle. # signs indicate power outage the day of the 6-h advance. (B) Summary of delay jetlag. (C) Summary of advance jetlag. n = 13 control and 14 dMHbCKO mice. (D) Summary of phase angle of entrainment. n = 23 control and 28 dMHbCKO mice. *p < 0.05, significant difference between genotypes.
dMHbCKO Mice Have Normal Expression of Clock Genes in the SCN
We have shown that reduced WRA in dMHbCKO mice is likely to result from a lack of motivation or reinforcement rather than a motor deficit that prevents wheel running (Hsu et al., 2014). However, the possibility remains that the reduced amplitude in circadian WRA of dMHbCKO mice results from a master circadian pacemaker that, because of reduced locomotor activity feedback, exhibits lower amplitude oscillation in its core molecular components. To test this hypothesis, we looked at the circadian expression of clock genes Per1 and Per2 by in situ hybridization in SCN slices from control and dMHbCKO mice sacrificed every 4 h over a 24-h period under DD conditions. The expression of mPer1 was rhythmic in both control and dMHbCKO mice and oscillated with a peak between CT4 and CT8 (interaction: F(5,38) = 1.23, p = 0.32; genotype: F(2,38) = 1.22, p = 0.28; time: F(5,38) = 23.31, p < 0.0001; Fig. 4A, C). A similar pattern was observed for mPer2 but with approximately a 4-h delay (interaction: F(5,38) = 1.53, p = 0.21; genotype: F(1,38) = 0.70, p = 0.41; time: F(5,38) = 19.58, p < 0.0001; Fig. 4B, D). We concluded that clock gene expression in the SCN is not affected by the dMHb lesion.
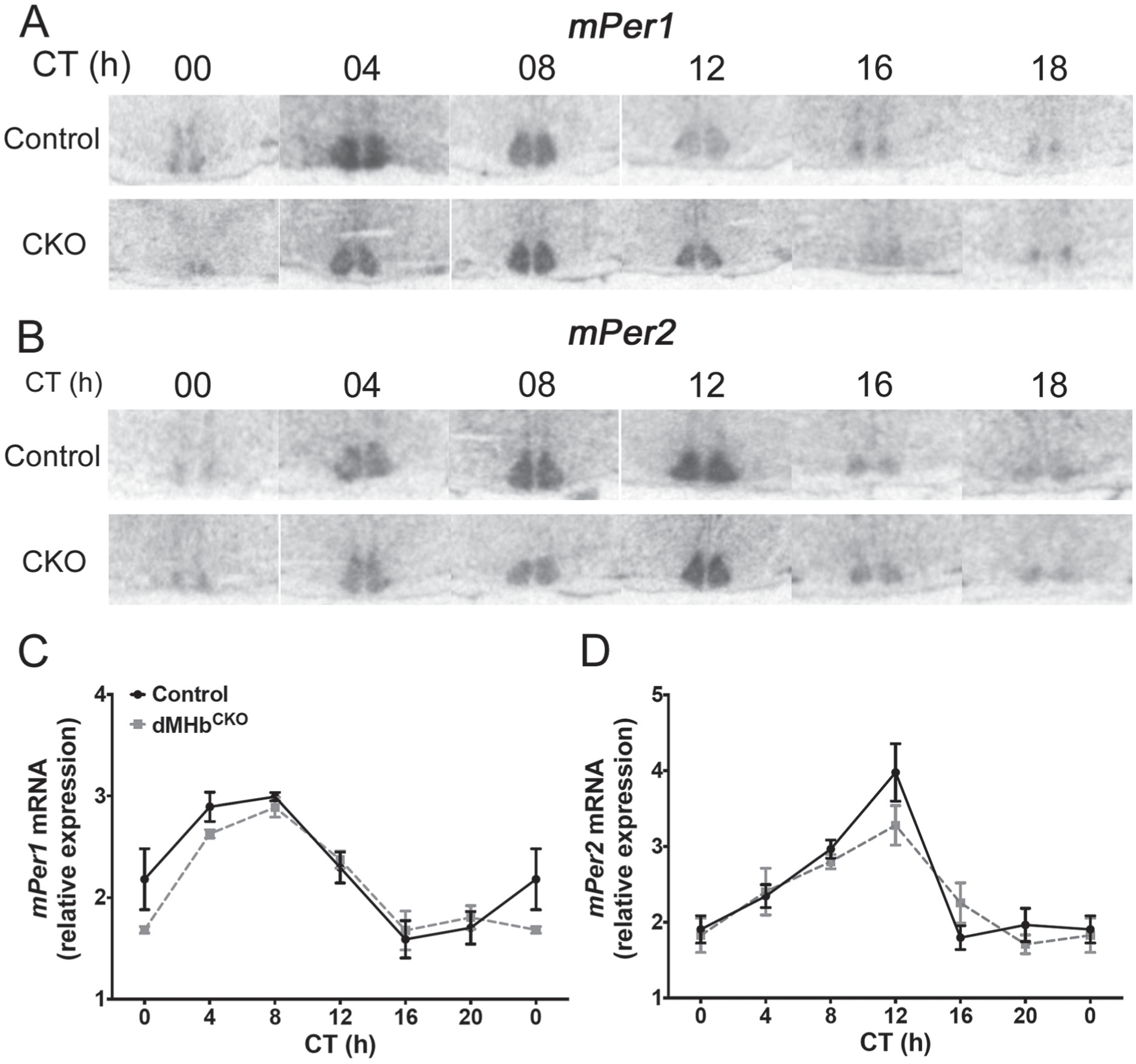
dMHbCKO mice exhibit normal circadian expression of mPer1 and mPer2 in the suprachiasmatic nucleus (SCN). (A, B) Representative autoradiographs of SCN sections from control and dMHbCKO mice sacrificed at different circadian time points and hybridized with a radiolabeled antisense probe against mPer1 (A) or mPer2 (B). (C, D) Normalized optical densities from control and dMHbCKO SCN slices hybridized against mPer1 (C) or mPer2 (D). Values for ZT0 are replotted at the end for easier visualization. Two-way analysis of variance yielded a significant effect of time but no significant effect of genotype or interaction. n = 3 to 5 animals per time point.
Discussion
The present study confirms our previous findings that dMHb-lesioned mice exhibit reduced voluntary WRA under a 12:12 LD cycle (Hsu et al., 2014) and shows that the phenotype persists under constant darkness. In contrast, the lesion has virtually no effect on HCA measured under either 12:12 LD or constant conditions. These results strengthen our interpretation that dMHb neurons are essential for the reinforcement of wheel running and suggest that their ablation has no effect on the amplitude of the circadian pacemaker driving locomotor activity. This interpretation is consistent with our finding that dMHbCKO mice have normal expression of Per1 and Per2 in the SCN and normal 24-h temporal regulation of sleep. dMHbCKO mice exhibit a longer circadian period than control mice for WRA but not for HCA. Intense activity such as WRA can induce both phase shifts and shortening of the free-running period of mice (Edgar et al., 1991; Yamada et al., 1988). Thus, our results suggest that the lower levels of running-wheel activity, which result from reduced motivation in dMHbCKO mice, fail to exert the normal levels of locomotor activity feedback necessary to change the period of the central circadian pacemaker. In contrast, reduced wheel running does not seem to affect the amplitude of the pacemaker, as rhythmic clock gene expression within the SCN remained unaltered in dMHbCKO mice.
The longer circadian period found in dMHbCKO mice was consistent with the fact that they reentrained faster, relative to control mice, in response to an abrupt delay of the LD cycle. In contrast, dMHbCKO mice did not show slower reentrainment to abrupt advances of the LD cycle. This apparent paradox may be due to the fact that entrainment of the circadian system by advances appears to rely more on the discrete effects of light on the phase of the circadian pacemaker whereas entrainment by delays more on the continuous effect of light on the pacemaker period (Schwartz et al., 2011).
The longer circadian period of WRA in dMHbCKO mice is consistent with lengthening of circadian period in hamsters bearing FR transections (Paul et al., 2011) and suggests that the effects of FR lesion may be specifically due to the destruction of dMHb efferent projections. However, Paul et al. (2011) also found that FR transection increased the HCA circadian period, which was unaffected by our dMHb lesion. Furthermore, whereas FR transection led to a decrease in the amount of WRA, it had the opposite effect on HCA (Paul et al., 2011). Because FR transections sever axons emerging from neurons in both the lateral and medial habenula, discrepancies between FR transection and genetic ablation of the dMHb suggest that the 2 subregions in the habenula may have different roles on the regulation of WRA, HCA, and their circadian regulation. However, species differences between hamsters and mice in habenula function cannot be excluded.
We believe that reduced WRA in dMHbCKO mice reflects a decrease in motivation rather than a motor deficit, as dMHbCKO mice also display a reduction in sucrose preference, a model of anhedonia (Hsu et al., 2014). It seems that the dMHb is required to convey multiple types of reinforcement and in turn for the maintenance of mood. This interpretation is consistent with the observation that wheel running is an elective behavior that is even displayed by mice living in their natural environment (Meijer and Robbers, 2014). The role of the dMHb in conveying reinforcing signals is further supported by intracranial self-stimulation experiments in which dMHb neurons are selectively activated using optogenetics (Hsu et al., 2014). In contrast, in tests of “behavioral despair” such as the forced swim test and tail suspension test, dMHbCKO mice do not show depression-like behavior (Hsu et al., 2016; Hsu et al., 2014). Here, we show that reduced WRA is associated with lengthening of the circadian period. Moderate lengthening of the circadian pacemaker period can lead to a prominent delay in the phase of entrainment (Johnson et al., 2003), which in turn has been associated with depression (Emens et al., 2009; Lewy et al., 2006). The phase angle of entrainment in dMHbCKO mice was not statistically different from that of controls, but it did show more variability. In summary, the dMHb contribution to mood stability that we propose likely relies on its ability to reinforce rewarding stimuli and does not appear to be associated with a delayed phase of entrainment. Finally, the resilience of dMHbCKO mice in some models of depression-like behavior (Hsu et al., 2016; Hsu et al., 2014) also supports the idea that these models do not reflect a single functional pathway.
Importantly, while dMHb ablation does not seem to affect the daily timing of sleep, it leads to increased sleep fragmentation and increased levels of REM sleep. Both sleep fragmentation and prolonged REM sleep bouts that lead to an overall increase in REM sleep are signature sleep symptoms in depressed patients (Palagini et al., 2013). Cui and collaborators (2014) have recently shown that mice with increased activity of lateral habenula (LHb) neurons, after local genetic KO of the glutamate transporter GLT-1, show both a depressive phenotype and an increase in REM sleep. Furthermore, lesions of the FR (Valjakka et al., 1998) or of the LHb (Aizawa et al., 2013) lead to deficits in REM sleep. dMHbCKO mice display phenotypic traits of depression including lack of motivation for wheel-running behavior and anhedonia, sleep fragmentation, and increased REM sleep. Thus, the different, and sometimes opposing, roles that are emerging for the LHb and the dMHb are not just limited to the regulation of reinforcement of rewarding stimuli (Hsu et al., 2014) and circadian modulation of locomotor activity but also to the regulation of sleep.
The neural pathways by which the dMHb increases motivation for WRA, and in turn changes in the circadian period, remain to be determined. Nonphotic stimuli, including WRA, that modulate the phase and period of circadian rhythms can exert their effect through direct projections from the thalamic intergeniculate leaflet of the thalamus and the serotonergic raphe nucleus (RN) to the SCN (Yannielli and Harrington, 2004). Whereas both tract-tracing and physiological studies have characterized a direct projection from the LHb to the RN (Aghajanian and Wang, 1977; Kalen et al., 1989; Nishikawa and Scatton, 1985; Wang and Aghajanian, 1977), the connectivity between the dMHb—its principal target the lateral interpeduncular nucleus—and centers regulating mood, circadian, and sleep physiology remain to be determined.
Footnotes
Acknowledgements
This work was supported by National Institutes of Health (NIH) awards DA035838 and MH093667 to E.E.T and National Science Foundation award IOS0909716 and NIH award NS094211 to H.O.D. Y.-W.A.H. was supported by the National Institute of Mental Health Training award F32MH098498. J.J.G. was supported by the Gates Millennium Scholarship, a Washington Research Foundation Fellowship, and a McNair Fellowship. J.G.P. was supported by the Mary Gates Undergraduate Fellowship and the Levinson and Sargent Awards. M.B.-H. was supported by the Israeli Committee for Higher Education Postdoctoral Fellowship for Women and by the Washington Research Foundation Funds for Innovation in Neuroengineering. BAC transgenic mice were generated by the Gene Expression Nervous System Atlas (GENSAT) Project, NINDS contracts N01NS02331 and HHSN271200723701C to The Rockefeller University (New York, NY). The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding agencies.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.