
Abstract
Circadian clocks are cell autonomous, transcriptionally based, molecular mechanisms that confer the selective advantage of anticipation, enabling cells/organs to respond to environmental factors in a temporally appropriate manner. Critical to circadian clock function are 2 transcription factors, CLOCK and BMAL1. The purpose of the present study was to reveal novel physiologic functions of BMAL1 in the heart, as well as to determine the pathologic consequences of chronic disruption of this circadian clock component. To address this goal, we generated cardiomyocyte-specific Bmal1 knockout (CBK) mice. Following validation of the CBK model, combined microarray and in silico analyses were performed, identifying 19 putative direct BMAL1 target genes, which included a number of metabolic (e.g., β-hydroxybutyrate dehydrogenase 1 [Bdh1]) and signaling (e.g., the p85α regulatory subunit of phosphatidylinositol 3-kinase [Pik3r1]) genes. Results from subsequent validation studies were consistent with regulation of Bdh1 and Pik3r1 by BMAL1, with predicted impairments in ketone body metabolism and signaling observed in CBK hearts. Furthermore, CBK hearts exhibited depressed glucose utilization, as well as a differential response to a physiologic metabolic stress (i.e., fasting). Consistent with BMAL1 influencing critical functions in the heart, echocardiographic, gravimetric, histologic, and molecular analyses revealed age-onset development of dilated cardiomyopathy in CBK mice, which was associated with a severe reduction in life span. Collectively, our studies reveal that BMAL1 influences metabolism, signaling, and contractile function of the heart.
Marked time-of-day-dependent oscillations in cardiovascular function are observed in mammals, including increased blood pressure, heart rate, and cardiac output during the awake period (Degaute et al., 1994; Degaute et al., 1991; Delp et al., 1991; Richards et al., 1986). Adverse cardiovascular events (e.g., myocardial infarction, sudden cardiac death) also exhibit a time-of-day-dependence in humans, with regard to their onset (Carson et al., 2000; Muller et al., 1989), while various behaviors known to adversely affect normal daily rhythms (e.g., night shift work) significantly increase cardiovascular disease risk (Harma and Ilmarinen, 1999; Knutsson et al., 1986; Koller 1983). A host of neurohumoral factors oscillate over the course of the day (e.g., autonomic, adrenergic, and sympathetic stimulation), many of which likely contribute to daily rhythms in physiologic and pathologic cardiovascular parameters (Muller et al., 1989; Prinz et al., 1979; Richards et al., 1986). However, it is becoming increasingly clear that cardiovascular components exhibit time-of-day–dependent oscillations in their responsiveness to extrinsic factors, which in turn likely influence outcomes following pathologic events (e.g., myocardial infarction; Collins and Rodrigo, 2010; Durgan et al., 2010, 2006, 2011b; Durgan and Young, 2010; Sachan et al., 2011). Currently, the relative contribution of extrinsic versus intrinsic factors toward rhythms in cardiovascular function and dysfunction remains undefined. Recently, multiple studies by Scheer and Shea highlighted that time-of-day–dependent oscillations in epinephrine, norepinephrine, fibrinolytic activity, heart rate, and blood pressure are mediated, at least in part, by an intrinsic circadian system in humans (Scheer et al., 2010, 2011; Shea et al., 2011). One intrinsic mechanism that has emerged as a significant modulator of the responsiveness of cardiovascular-relevant cells is the circadian clock (Rudic and Fulton, 2009; Young, 2009).
Circadian clocks are cell autonomous molecular mechanisms that confer the selective advantage of anticipation, facilitating responsiveness of a cell/organ to an extrinsic stimulus/stress in a temporally appropriate manner (Edery, 2000). The circadian clock is a transcriptionally based mechanism that has been identified in all mammalian cells investigated to date (Dibner et al., 2010; Takahashi et al., 2008). At the core of the mechanism are 2 transcription factors, CLOCK and BMAL1, which, upon heterodimerization, bind to E-boxes in the promoters of target genes (Gekakis et al., 1998; Hogenesch et al., 1998). Ubiquitous genetic disruption of circadian clock genes, as well as clock-controlled genes, often precipitates cardiovascular dysfunction. For example, heterozygous tau hamsters (that harbor a mutation in CK1ϵ, resulting in a 22-h circadian clock) develop hypertrophic cardiomyopathy (Martino et al., 2008). A cardiomyopathic phenotype is also observed when the clock output genes Dbp, Hlf, and Tef are genetically deleted (Wang et al., 2010). Interestingly, targeted ablation of the core clock component Bmal1 in a ubiquitous manner results in severe cardiac dysfunction, which is associated with increased mortality (Kondratov et al., 2006; Lefta et al., 2012). Collectively, these observations provide evidence that functional circadian clocks are essential for normal cardiovascular function.
In an attempt to define roles for the cardiomyocyte-specific circadian clock, we have previously targeted the transcription factor CLOCK in a dominant negative manner. Using cardiomyocyte-specific Clock mutant (CCM) mice, we have highlighted critical roles for this transcription factor in both cardiac physiology (e.g., transcription, metabolism, contractile function) and pathophysiology (e.g., ischemia/reperfusion tolerance, hypertrophic growth; Bray et al., 2008; Durgan et al., 2006, 2010, 2011a, 2011b; Tsai et al., 2010). In contrast, the role of BMAL1 in the cardiomyocyte remains undefined. To address this deficiency, we recently generated a cardiomyocyte-specific Bmal1 knockout (CBK) mouse model (Durgan, et al., 2011b). Through combined microarray and in silico analyses, the present study identified 19 putative direct BMAL1 target genes in the hearts, many of which influence metabolism and signaling. Validation studies confirmed that Bdh1 and Pik3r1 are regulated by BMAL1 and that their chronic repression in CBK hearts is associated with impaired ketone body metabolism and signaling, respectively. In addition, CBK hearts exhibit abnormalities in glucose utilization in the fed state as well as abnormal metabolic responses to acute fasting (i.e., a physiologic metabolic stress). CBK mice also exhibit an age-onset cardiomyopathy, which is associated with early mortality. Collectively, these observations highlight critical roles of BMAL1 in the heart.
Materials and Methods
Mice
The present study used 1) CBK (BMAL1flox/flox/α-MHC-CRE+/−) and littermate controls (BMAL1flox/flox/α-MHC-CRE−/−) on the C57Bl/6J background, 2) α-MHC-CRE (α-MHC-CRE+/−) and littermate wild-types (α-MHC-CRE−/−) on the C57Bl/6J background, and 3) CCM (α-MHC-dnCLOCK+/−) and littermate wild-types (α-MHC-dnCLOCK−/−) on the FVB/N background; both CBK and CCM mice have been described previously (Durgan et al., 2006, 2011b). All experimental mice were male and were housed at the Center for Comparative Medicine at the University of Alabama at Birmingham (UAB), under temperature-, humidity-, and light-controlled conditions. A strict 12-h light/12-h dark cycle regime was enforced (lights on at 0600 h; zeitgeber time [ZT] 0); the light/dark cycle was maintained throughout these studies, facilitating elucidation of the potential roles for cardiomyocyte circadian clock components under physiological conditions. As such, diurnal variations were investigated in mice (as opposed to circadian rhythms). Mice received food and water ad libitum, unless otherwise stated. Mice were housed in standard micro-isolator cages, with the exception of the whole-body behavioral/metabolic analyses and fasting studies, during which time mice were housed within Comprehensive Laboratory Animal Monitoring System (CLAMS) and wire-bottom cages, respectively. All animal experiments were approved by the Institutional Animal Care and Use Committee of the UAB.
Whole-Body Behavioral and Metabolic Monitoring
Twenty-four hour patterns of physical activity, food intake, and energy expenditure (indirect calorimetry) were measured in mice using a CLAMS (Columbus Instruments Inc., Columbus, OH), as described previously (Bray et al., 2013).
Echocardiographic Analysis
Left ventricular function was determined in vivo through echocardiography, at an established UAB core facility. Mice were anesthetized using 1.5% isofluorane with 95% oxygen; heart rate and body temperature were kept constant. M-mode echocardiography was performed using the Visualsonics imaging/processing system as described previously (Bray et al., 2008). Control versus CBK assessments were performed in 2011 to 2012 on a Vevo 2100 (Toronto, Canada), whereas wild-type versus α-MHC-Cre assessments were performed in 2013 by a different technician on a Vevo 7700. As such, direct comparisons are possible only within each distinct substudy.
Humoral Factor Analysis
Plasma glucose, triglyceride, nonesterified fatty acids (NEFA), insulin, leptin, and adiponectin concentrations were measured with commercially available kits (Stanbio Laboratory, Boerne, TX; EMD Millipore, Billerica, MA; TOSOH Bioscience, San Francisco, CA; Wako Chemicals, Richmond, VA) using a Sirrus Clinical Chemistry Analyzer (Stanbio Laboratory, Boerne, TX).
Histologic Assessment
Cross sections from the medial heart were taken immediately upon removal of heart and flash frozen in OCT. Laminin staining was used for measurement of myocyte cross-sectional area; at least 100 myocytes were assessed per heart using ImagePro Plus software (Media Cybernetics, Inc., Rockville, MD), as described previously (Durgan et al., 2011b). Picrosirius Red staining of collagen fibers was used for semi-quantitative measurement of left ventricular fibrosis, using ImagePro Plus software (Media Cybernetics, Inc.), as described previously (Durgan et al., 2011b).
Quantitative RT-PCR
RNA was extracted from hearts using standard procedures (Chomczynski and Sacchi, 1987). Candidate gene expression analysis was performed by quantitative reverse transcription polymerase chain reaction (RT-PCR), using previously described methods (Gibson et al., 1996; Heid et al., 1996). For quantitative RT-PCR, specific Taqman assays were designed for each gene from mouse sequences available in GenBank. Primer and probe sequences used are listed in Supplemental Table S1. All quantitative RT-PCR data are presented as fold-change from littermate control.
Microarray Analysis
Microarray analysis was performed using the mouse Ref-8 BeadChips and the BeadStation System (Illumina Inc., San Diego, CA) according to the manufacturer’s guidelines, as described previously (Bray et al., 2008). Ventricular tissue was collected from CBK and littermate controls every 3 h for a period of 24 h (n = 4 samples per time point; 64 samples total), and RNA was extracted. Genes/transcripts were defined as being significantly expressed above background when the 75th percentile of each gene’s detection score met or exceeded 0.9. The expression data were normalized within centiles of the distribution of gene expression values. Time-of-day–independent differentially expressed genes between CBK and littermate control samples were identified by 2-way analysis of variance (ANOVA; i.e., genotype main effect). Time-of-day–dependent differential expression was performed by initially identifying genes exhibiting significant oscillations in control samples using cosinor analysis, as described previously (Bray et al., 2008); those genes that oscillate in control samples and either do not oscillate in CBK samples or oscillated with a decreased amplitude (assessed by 2-way ANOVA) were identified as being differentially expressed in a time-of-day–dependent manner. Normalized data have been submitted to the GEO archive and are available at http://www.ncbi.nlm.nih.gov/geo/ (GSE43073).
To identify a comprehensive set of putative direct BMAL1 gene targets, we performed a systems-level bioinformatics analysis. To do this, we compared the most highly induced and repressed genes identified from our CBK microarray data (using fold-change cutoffs of 2.0 and 0.5, respectively, for the time-of-day–independent differentially expressed genes; Suppl. Table S3) against a genome-wide map of murine BMAL1 binding targets identified through functional assays by chromatin immunoprecipitation (ChIP) coupled with deep sequencing (Rey et al., 2011). In addition, we performed an in silico search within the 5′-regulatory regions of identified genes for putative circadian E-box sequences (CANNTG), the canonical palindromic circadian E-box motif (CACGTG), and E-box like sequences (E′-box) (CACGTT), as described previously (Tsimakouridze et al., 2012). The 1000 base-pair regions upstream of the coding sequences were retrieved from UCSC Genome Bioinformatics (http://genome.ucsc.edu/cgi-bin/hgTables). Moreover, we interrogated the circadian section of the mammalian Promoter/Enhancer DataBase ( http://promoter.cdb.riken.jp/circadian_home.html; Kumaki et al., 2008) for genes containing high-scoring putative E-boxes conserved in the noncoding region of both the human and mouse.
Western Blotting
Qualitative analysis of protein expression was performed using standard Western blotting techniques, as described previously (Durgan et al., 2011a). Lysates (10 µg) were separated on a 7.5% bis-acrylamide gel by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a PVDF membrane, and probed with anti-BMAL1 (Millipore; catalog No. AB2298), anti-BDH1 (Abcam, Cambridge, MA; catalog No. AB93931), anti-PIK3R1 (Abcam; catalog No. AB22653), anti-AKT (Millipore; catalog No. 05-591), anti-phospho-Thr308-AKT (Cell Signaling, Danvers, MA; catalog No. CS-9275), anti-GSK3β (Santa Cruz Biotechnology, Santa Cruz, CA; catalogue No. sc-7291), anti-phospho-Ser9-GSK3β (Cell Signaling; catalogue No. CS-9336), or GAPDH (Cell Signaling; catalogue No. CS-2118) antibodies. Membranes were incubated with either goat anti-rabbit or anti-mouse horse radish peroxidase–conjugated secondary antibody (Santa Cruz; catalog Nos. sc-2004 and sc-2005, respectively). Bands were visualized with Immunstar Western C detection kit (Bio-Rad, Hercules, CA) on x-ray film, scanned and quantified using Scion Image version 4.0.3.2, and normalized to Calsequestrin (Abcam; catalog No. AB3516). For phosphorylation to total comparisons, membranes were incubated with either AlexaFluor 680–conjugated secondary donkey anti-rabbit (Invitrogen, Carlsbad, CA; catalog No. A10043) or IRDYE800-conjugated secondary donkey anti-mouse (LI-COR, Lincoln, NE; catalog No. 926-32212). Bands were visualized with the LI-COR Odyssey CLx Infrared Imager and quantified using Image Studio version 4.0.21 software.
Enzymatic Activities
β-Hydroxybutyrate dehydrogenase (BDH) and citrate synthase (negative control) activities were measured in freshly isolated hearts using previously described assays (Grinblat et al., 1986; Srere, 1969). BDH activity was assessed by following the conversion rate of NAD+ to NADH in the presence of β-hydroxybutyrate. Citrate synthase activity was assessed by following the conversion rate of 5,5′-dithiobis-(2-nitrobenzoic acid) to 2-nitro-5-thiobenzoate in the presence of oxaloacetate and acetyl-CoA. Enzymatic activities were normalized to protein content (determined by BCA assay kit; Pierce Biotechnology, Rockford, IL).
Working Mouse Heart Perfusions
Myocardial substrate utilization was measured ex vivo through isolated working mouse heart perfusions, as described previously (Bray et al., 2008; Durgan et al., 2011a; Tsai et al., 2010, 2013). All hearts were perfused in the working mode (nonrecirculating manner) for 40 min with a preload of 12.5 mm Hg and an afterload of 50 mm Hg. Standard Krebs-Henseleit buffer was supplemented with 8 mM glucose, 0.4 mM oleate conjugated to 3% BSA (fraction V, fatty acid–free; dialyzed), 10 µU/mL insulin (basal/fasting concentration), 2 mM β-hydroxybutyrate, 0.2 mM acetoacetate, 0.05 mM L-carnitine, and 0.13 mM glycerol. Metabolic flux was assessed through the use of distinct radiolabeled tracers: 1) [U-14C]-β-hydroxybutyrate (0.04 mCi/L; β-hydroxybutyrate oxidation), 2) [U-14C]-glucose (0.12 mCi/L; glycolysis, glucose oxidation), and 3) [9,10-3H]-oleate (0.067mCi/L; β-oxidation). Measures of cardiac metabolism (e.g., β-hydroxybutyrate oxidation and oxygen consumption) and function (i.e., cardiac power and rate pressure product) were determined as described previously (Bray et al., 2008; Durgan et al., 2011a; Tsai et al., 2010, 2013). At the end of the perfusion period, hearts were snap-frozen in liquid nitrogen and stored at −80 °C prior to analysis. Data are presented as steady-state values (i.e., values during the last 10 min of the perfusion protocol).
Isolated Adult Cardiomyocyte Studies
Viable adult mouse and rat cardiomyocytes were isolated as described previously (Durgan et al., 2005; Marsh et al., 2011; Shan et al., 2008). Adult mouse cardiomyocytes were lysed immediately, for subsequent Western blot analysis. Adult rat cardiomyocytes were cultured overnight (equilibration period) followed by challenge with 50% serum for a period of 2 h (i.e., serum shock); cells were terminated at 4-h intervals thereafter. We have previously reported circadian clock gene oscillations in these samples (Durgan et al., 2005).
DNA Precipitation Assays
DNA precipitation assays were performed as described previously (Yang and Chow, 2012). Briefly, COS cells were transfected with expression vectors for Bmal1 and Clock and subsequently harvested in Trition-lysis buffer (20 mM Tris [pH 7.4], 134 mM NaCl, 2 mM EDTA, 25 mM β-glycerophosphate, 2 mM NaPPi, 10% glycerol, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, and 10 µg/mL leupeptin). Double-stranded biotinylated oligonucleotides (−573 bp E box, 5′-Biotin-CCTGAGTGAGACACTTGGCAGAGAGCACCTGTACTCTGCGTT-3′; −619 bp E box, 5′-Biotin-GAGATTACATCACCTTCCTTAAAGCAGCTAGCAATGAGATG-3′; −1828 bp E box, 5′-Biotin-GGGCTGGGACCACTTGGTAGAAGAACAGCTTCTTTGAA-3′) were incubated with BMAL1 and CLOCK cell extracts for 12 h before precipitation with 20 µL of streptavidin-Sepharose for 2 h. After 3 washes in Triton-lysis buffer, precipitated DNA and its associated proteins were separated by SDS-PAGE, and an immunoblot was performed to detect BMAL1.
Luciferase Reporter Assays
The Pik3r1 promoter was amplified from mouse genomic DNA and subcloned into the pGL3 basic luciferase reporter plasmid using MluI and XhoI sites. Expression vectors for Bmal1 and Clock (50 ng) were co-transfected with the Pik3r1 luciferase reporter plasmids (100 ng) and control plasmid pRSV β-galactosidase (25 ng) into COS cells. Transfected cells were serum shocked (with 20% serum) for 2 h and subsequently maintained in media supplemented with 2% serum for 24 h before harvest. The data are presented as relative luciferase activity, calculated as the ratio of luciferase activity to the activity of β-galactosidase.
Statistical Analysis
Statistical analyses were performed using Student t tests, 2-way ANOVA, and cosinor analysis, as described previously (Bray et al., 2008, 2013). Briefly, Excel 2010 was used to compare CBK/CCM to littermate controls (only when 2 experimental groups were investigated). Stata version IC10.0 (Stata Corp., San Antonio, TX) or IBM SPSS Statistics 22.0 (IBM, Inc., Armonk, NY) was used to perform 2-way ANOVA (with repeated measures, when appropriate) to investigate main effects of genotype, time, feeding status, and/or age (indicated in the figures), followed by Bonferroni post hoc analyses for pairwise comparisons (only when a significant interaction was observed). Assumptions of normality and homogeneity of variances were met and verified with Shapiro-Wilk and Levene’s tests. For repeated-measures ANOVA, sphericity (for repeated measures) was examined with Mauchly’s test, and degrees of freedom were modified using the Greenhouse-Geisser adjustment when appropriate. Survival data were analyzed by using a Kaplan-Meier survival analysis with a log rank method of statistics. Cosinor analysis was conducted using nonlinear regression with Stata version IC10.0 and SPSS 22. The cosinor analyses were performed based on the assumption that the free-running period is fixed at 24 h. Data were considered rhythmic if the p value of the R2 of the linearized cosinor function of f(t) = M + Cos(2πt/24) + Sin(2πt/24) was less than 0.05 (indicated in Suppl. Table S2). In cases in which both comparison groups significantly fit the cosine function, individual parameter estimates for mesor, amplitude, and phase were compared using t tests determined from the pooled variances. In all analyses, the null hypothesis of no model effects was rejected at p < 0.05.
Results
Cardiomyocyte-Specific Bmal1 Ablation Selectively Affects the Circadian Clock in the Heart
To initially validate the CBK model, Bmal1 gene and protein expression were assessed. Figure 1Ai-ii shows decreased bmal1 gene (60%) and BMAL1 protein (67%) expression in intact hearts isolated from 12-wk-old CBK mice at ZT6 (relative to littermate controls). Importantly, BMAL1 protein was almost undetectable in cardiomyocytes isolated from CBK hearts (Fig. 1Aiii), suggesting that residual expression in intact CBK hearts is due primarily to the presence of noncardiomyocyte cell types (e.g., vascular smooth muscle cells, endothelial cells, fibroblasts). Decreased BMAL1 expression in CBK hearts was associated with a dramatic attenuation in time-of-day–dependent rhythmic oscillations for core circadian clock components (i.e., bmal1, rev-erbα, per1, and per3) and output (i.e., dbp and e4bp4) genes; in the case of clock output genes (considered a marker of clock function), the amplitude of dbp mRNA oscillations was decreased by 73% in CBK hearts, while those of e4bp4 mRNA were completely abolished (Fig. 1B; Suppl. Table S2). In contrast, circadian clock function in extracardiac peripheral tissues remained intact as expected, as evidenced by identical bmal1 and dbp mRNA oscillations in skeletal muscle (gastrocnemius), liver, kidney, and adipose (epididymal fat) isolated from CBK versus littermate control mice (Suppl. Fig. S1A; Suppl. Table S2). Similarly, no significant differences in humoral (i.e., glucose, triglyceride, leptin, adiponectin; Suppl. Fig. S1B; Suppl. Table S2) or whole-body behavioral (i.e., physical activity and food intake; Suppl. Fig. S1C; Suppl. Table S2) factor diurnal variations were observed between CBK and littermate control mice. Whole-body energy expenditure also did not significantly differ between CBK and littermate controls (Suppl. Fig. S1D; Suppl. Table S2). Collectively, these data are consistent with the cardiac-specific nature of the CBK model.
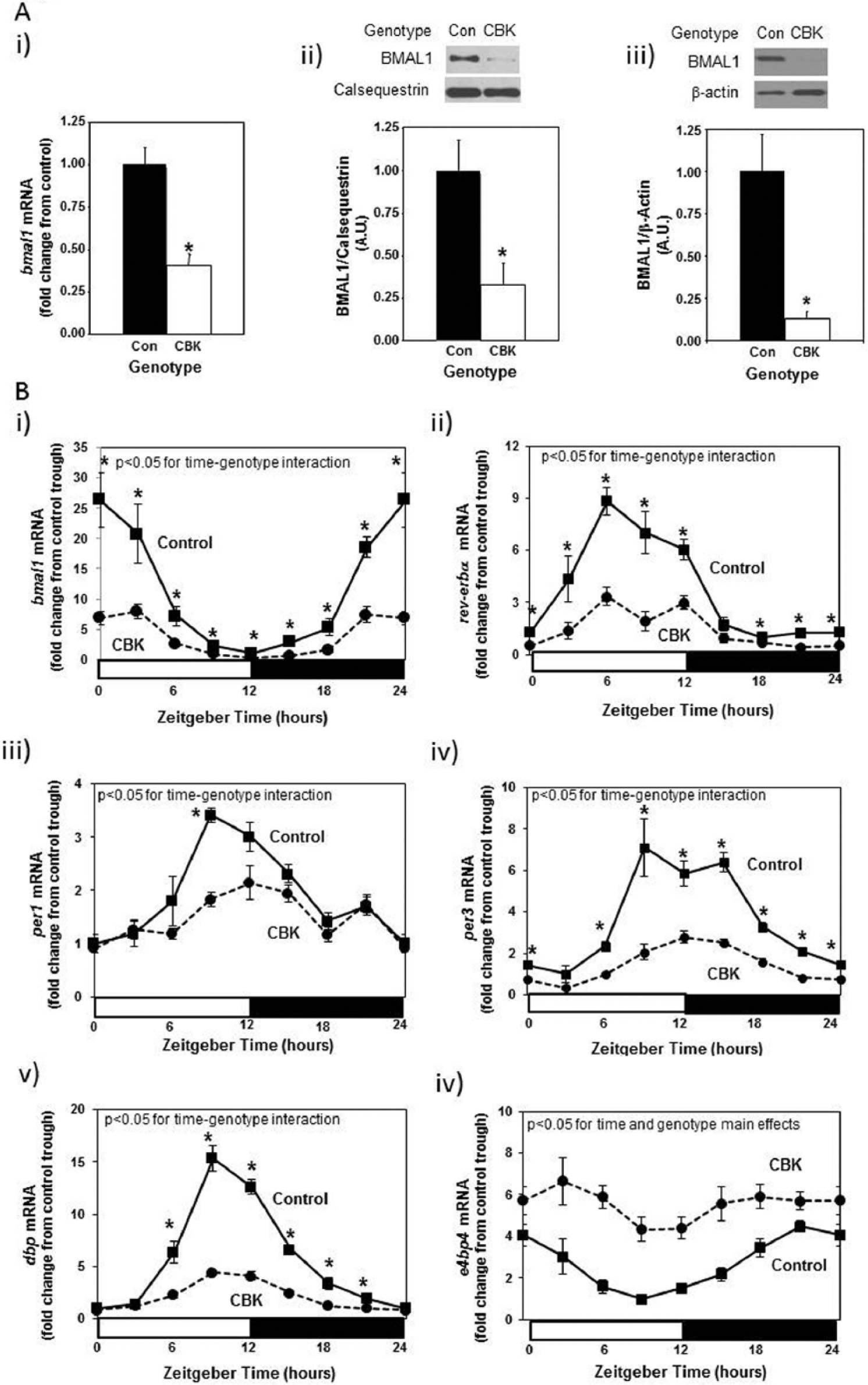
Decreased bmal1 mRNA levels in intact cardiomyocyte-specific Bmal1 knockout (CBK) hearts (Ai), decreased BMAL1 protein levels in intact CBK hearts (Aii) and in cardiomyocytes isolated from CBK hearts (Aiii), as well as attenuated time-of-day–dependent gene expression oscillations in circadian clock components (bmal1, rev-erbα, per1, and per3) and clock output genes (dbp and e4bp4) in CBK hearts (B). Mice were housed in a 12-h light:12-h dark cycle (lights on at ZT0). Hearts were isolated from 12-wk-old male CBK mice and age-matched littermate controls. For data presented in (A), hearts were isolated at ZT6. Note that ZT0 and ZT24 are identical data. Values are expressed as mean ± SEM (n = 4-12; sample size range varies dependent on the parameter investigated). *p < 0.05 for CBK versus littermate control at a distinct ZT (post hoc pairwise comparison).
Identification of Putative Direct BMAL1 Target Genes in the Heart
Given that BMAL1 is a transcription factor, we rationalized that investigating the impact of cardiomyocyte-specific genetic ablation of this protein on the cardiac transcriptome might identify novel roles for BMAL1 in the heart. Accordingly, ventricles were collected from 12-wk-old CBK and littermate control mice at 3-h intervals over the course of the 24-h light/dark cycle. Following RNA isolation, an unbiased microarray approach was taken. Microarray data were analyzed in 2 manners. First, genes chronically induced or repressed in a time-of-day–independent manner in CBK hearts, relative to their littermate controls, were identified through ANOVA; a total of 2037 genes were identified through this analysis, of which 1002 were induced while 1035 were repressed in CBK hearts (Suppl. Table S3). Second, genes that significantly oscillate in a time-of-day–dependent manner (with a periodicity of approximately 24 h) in control hearts, and that the amplitudes of these oscillations were significantly attenuated/abolished in CBK hearts, were identified through cosinor analysis; a total of 1267 genes were identified by this analysis (Suppl. Table S4). Figure 2 illustrates attenuation (i.e., decreased amplitude) and/or abolition of gene expression oscillations in CBK, relative to littermate control, hearts. It is interesting to note that CBK hearts appear to exhibit a similar temporal suspension as previously described in CCM hearts (Bray et al., 2008), wherein genes that oscillate in control hearts are generally suspended at the beginning of the light/sleep phase in CBK hearts (Fig. 2). Collectively, these observations suggest that genetic deletion of BMAL1 in the cardiomyocyte results in marked alterations in the cardiac transcriptome, in both time-of-day–independent and –dependent manners.
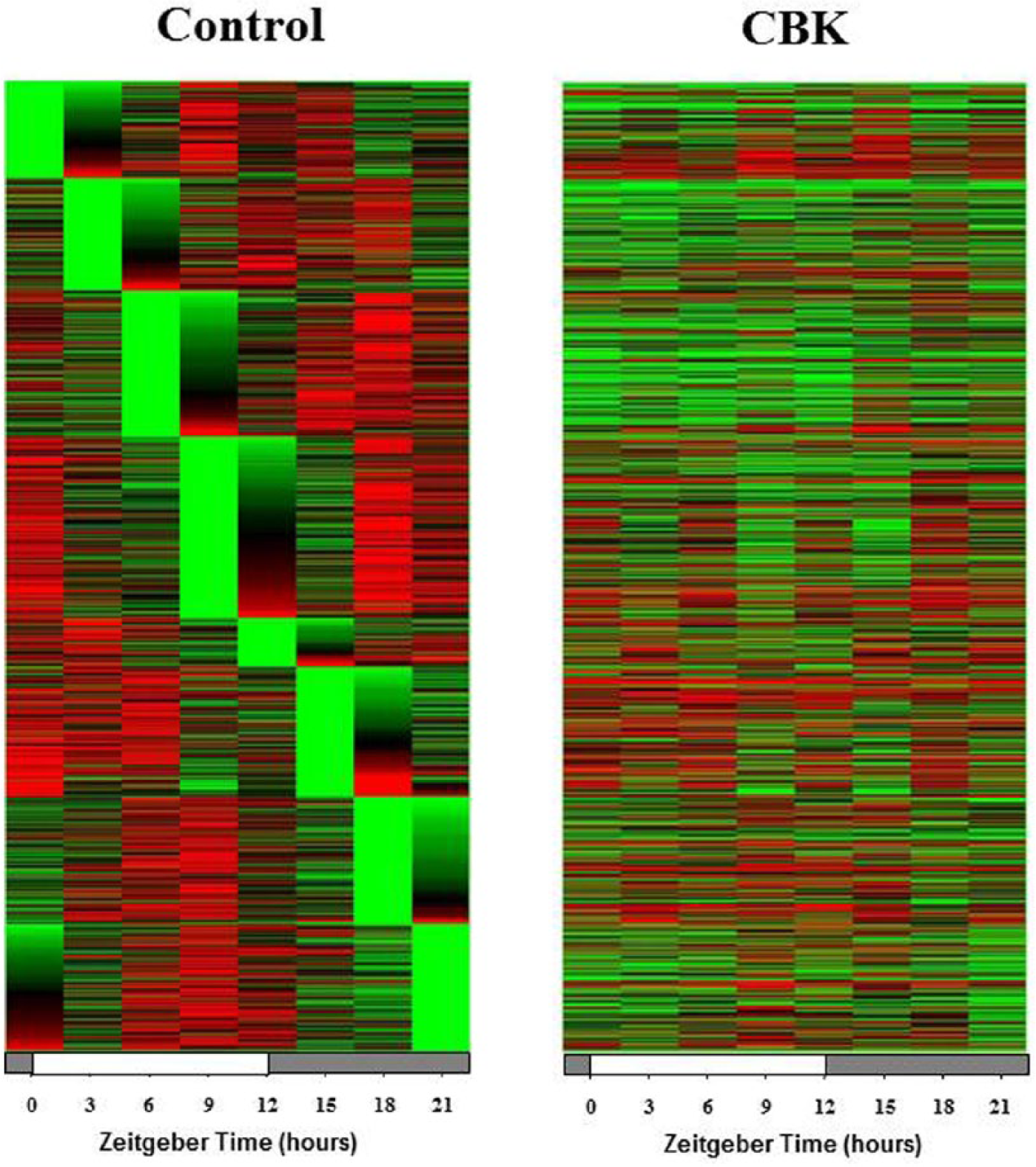
Heat map illustrating genes that oscillate in a time-of-day–dependent manner in control hearts (periodicity of 24 h), (i) while oscillations of these genes are significantly attenuated/abolished in cardiomyocyte-specific Bmal1 knockout hearts (ii). Green represents a low level of expression; red represents a high level of expression. Data are taken from Supplemental Table S5 (n = 4).
Next, in an attempt to identify putative candidate genes for which BMAL1 directly regulates in the heart, we systematically compared our microarray gene expression data to systems-level transcriptional regulatory databases (Kumaki et al., 2008; Rey et al., 2011; Tsimakouridze et al., 2012). A gene was defined as a putative direct BMAL1 target gene if it 1) was highly induced (fold-change of 2 or more) or repressed (fold-change of 0.5 or less) in CBK hearts; 2) the 5′-regulatory region of the gene contained a putative BMAL1/CLOCK binding site (i.e., E-box sequences [CANNTG], the canonical palindromic circadian E-box motif [CACGTG], or E-box–like sequences [CACGTT]); and 3) the gene was included in a genome-wide map of BMAL1 binding targets in mouse livers identified through functional assays by chromatin immunoprecipitation coupled with deep sequencing (Rey et al., 2011). It should be noted that the latter step likely underestimates the number of BMAL1 target genes in the heart, as previous microarray studies have suggested a high level of tissue specificity in time-of-day–dependent regulation of gene expression (Storch et al., 2002). This analysis revealed a total of 19 putative direct BMAL1 target genes (Table 1), 16 of which were previously identified as CLOCK-regulated genes through transcriptome analysis of CCM hearts (Bray et al., 2008); of these 16 genes, 15 (i.e., 94%) were differentially expressed in CCM and CBK hearts in an identical manner. Among the 19 putative BMAL1 target genes, the expression of 9 oscillate significantly in wild-type hearts with a periodicity of approximately 24 h (while 10 do not; Table 1). Based on known biologic function, these 19 genes appeared to fall into 3 main categories, namely, clock function, metabolism, and signaling (Table 1).
Putative direct BMAL1 target genes in the heart.
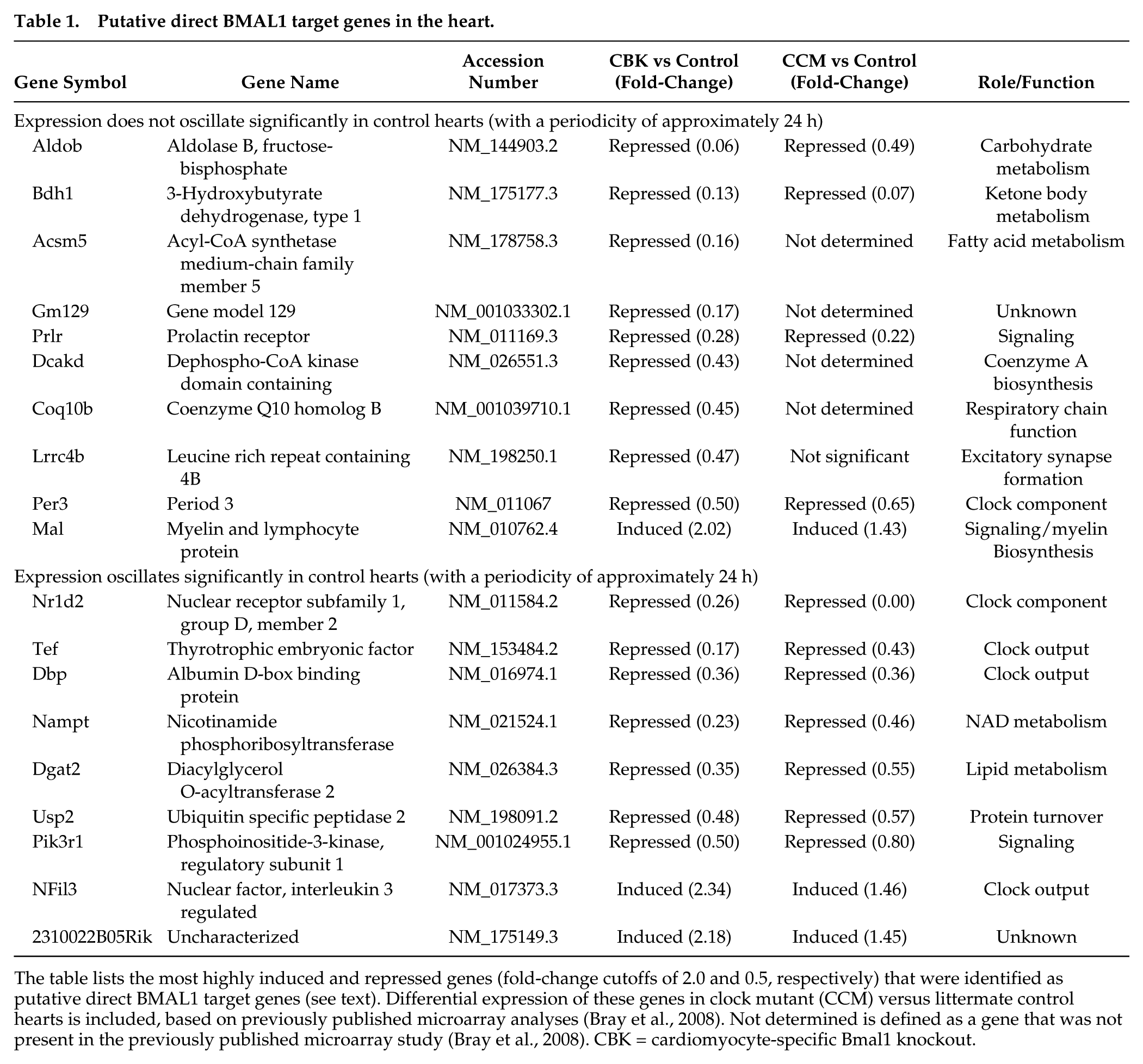
The table lists the most highly induced and repressed genes (fold-change cutoffs of 2.0 and 0.5, respectively) that were identified as putative direct BMAL1 target genes (see text). Differential expression of these genes in clock mutant (CCM) versus littermate control hearts is included, based on previously published microarray analyses (Bray et al., 2008). Not determined is defined as a gene that was not present in the previously published microarray study (Bray et al., 2008). CBK = cardiomyocyte-specific Bmal1 knockout.
BMAL1 Regulates Bdh1 in a Time-of-Day–Independent Manner
We next sought to validate our combined microarray and in silico analyses. Through the use of CCM mice, we have previously reported that 2 of the putative direct BMAL1 target genes listed in Table 1 that are known to influence metabolism (namely, Dgat2 and Nampt) are regulated by the cardiomyocyte circadian clock (Durgan et al., 2011a; Tsai et al., 2010). One metabolism-related gene identified in Table 2 that has not been investigated previously, in terms of circadian clock regulation, encodes for β-hydroxybutyrate dehydrogenase 1 (Bdh1). RT-PCR validation confirmed that bdh1 mRNA levels in CBK hearts (relative to wild-type littermates) were decreased (83%) in a time-of-day–independent manner (Fig. 3Ai-ii; Suppl. Table S2). Decreased bdh1 expression was not due to the expression of Cre alone, as α-MHC-Cre mouse hearts exhibit normal bdh1 mRNA levels (1.00 ± 0.17 versus 0.94 ± 0.26 A.U. for α-MHC-Cre and wild-type littermate hearts isolated at ZT15, respectively; p > 0.05). bdh1 mRNA levels are also significantly reduced (94%) in CCM hearts in a time-of-day–independent manner (Fig. 3Aiii; Suppl. Table S2). Consistent with the lack of significant oscillation of bdh1 mRNA levels in control hearts, this transcript did not significantly oscillate in synchronized adult rat cardiomyocytes (Fig. 3B; p > 0.05 for cosine analysis). BDH1 protein levels also did not oscillate in a time-of-day–dependent manner in control hearts (although BDH1 protein levels are lower in both CBK [87%] and CCM [85%] hearts; Fig. 3C; Suppl. Table S2). Interestingly, decreased BDH1 protein levels in CBK and CCM hearts are associated with decreased BDH enzymatic activity (95% and 91% decrease, respectively; Fig. 3D), in the absence of genotype-dependent alterations in citrate synthase activity (Suppl. Fig. 2A). Similarly, β-hydroxybutyrate oxidation rates were decreased in ex vivo perfused CBK and CCM hearts (61% and 67% decrease, respectively; Fig. 3E), in the absence of genotype-dependent alterations in myocardial oxygen consumption or contractile function (Suppl. Fig. S2B). Note that BDH enzymatic activity and β-hydroxybutyrate oxidation rates were assessed at only one time of the day (ZT6), as BDH1 protein levels do not oscillate. Collectively, our findings suggest that Bdh1 is likely influenced by both BMAL1 and CLOCK, in a time-of-day–independent manner.
Gravimetric, histologic, and transcriptional measures in both 12- and 36-wk-old CBK and littermate control mice.
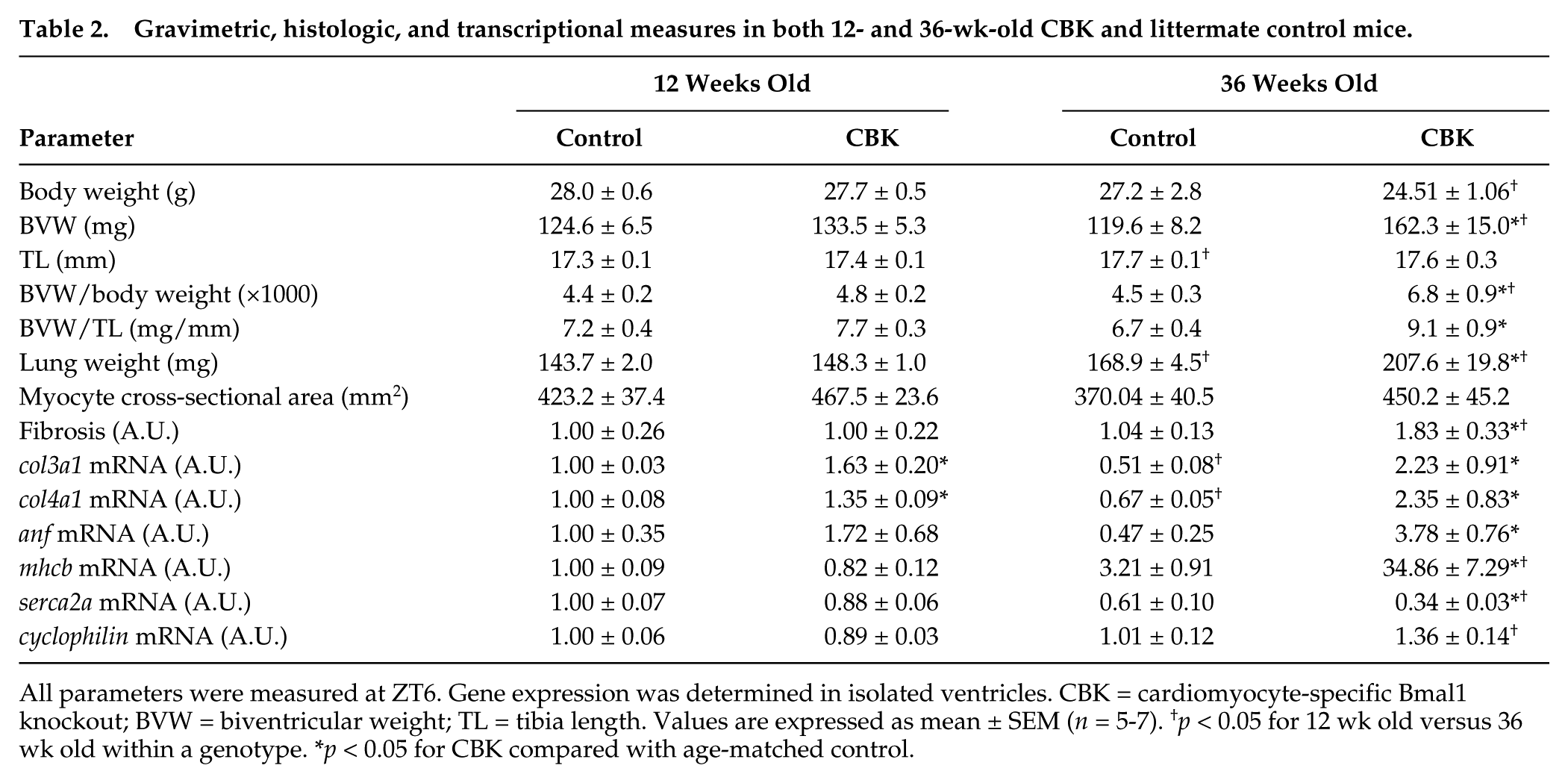
All parameters were measured at ZT6. Gene expression was determined in isolated ventricles. CBK = cardiomyocyte-specific Bmal1 knockout; BVW = biventricular weight; TL = tibia length. Values are expressed as mean ± SEM (n = 5-7). †p < 0.05 for 12 wk old versus 36 wk old within a genotype. *p < 0.05 for CBK compared with age-matched control.
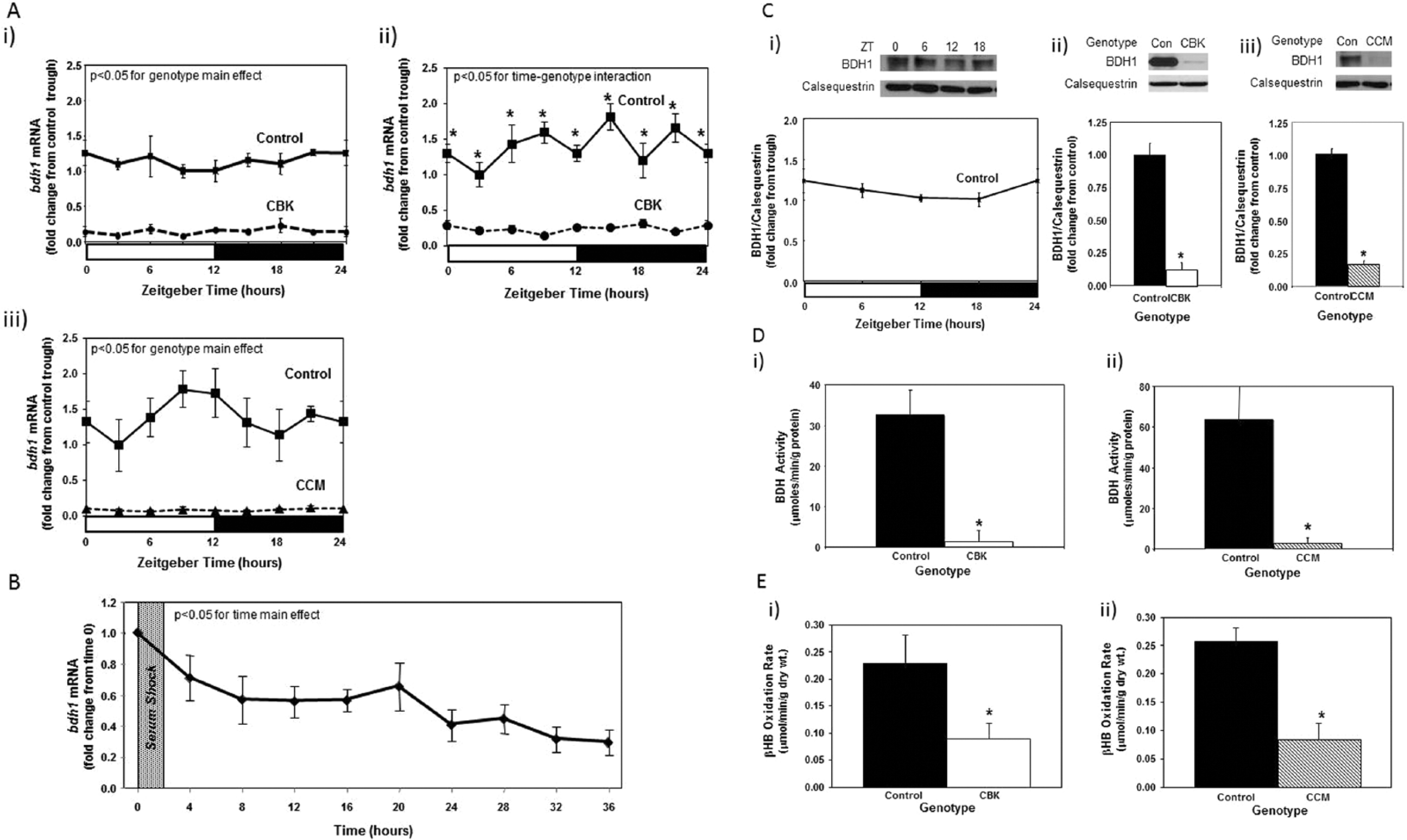
Alterations in bdh1 mRNA in cardiomyocyte-specific Bmal1 knockout (CBK; microarray [i], RT-PCR [ii]) and Clock mutant (CCM; RT-PCR [iii]) hearts, compared with littermate controls (A). Lack of bdh1 mRNA oscillations in serum shocked adult rat cardiomyocytes (i.e., challenged with 50% serum for 2 h [shaded area]; B). Lack of diurnal variations in BDH1 protein levels in control hearts (Ci). Decreased BDH1 protein levels in CBK (Cii) and CCM (Ciii) hearts at ZT6. Decreased β-hydroxybutyrate dehydrogenase activity in CBK (Di) and CCM (Dii) hearts at ZT6. Decreased β-hydroxybutyrate oxidation in ex vivo perfused CBK (Ei) and CCM (Eii) hearts at ZT6. Mice were housed in a 12-h light:12-h dark cycle (lights on at ZT0). Hearts were isolated from 12-wk-old male CBK, CCM, and age-matched littermate controls. Note that ZT0 and ZT24 are identical data. Values are expressed as mean ± SEM (n = 3-7; sample size range varies dependent on the parameter investigated). *p < 0.05 for CBK/CCM versus littermate control at a distinct ZT (post hoc pairwise comparison).
BMAL1 Regulates Pik3r1 in a Time-of-Day–Dependent Manner
An additional putative BMAL1 target gene identified in Table 1 is Pik3r1, encoding for the p85α regulatory subunit of phosphatidylinositol 3-kinase (PI3K). This gene is of potential interest because 1) it is a component of the insulin signaling cascade, which affects both cardiac metabolism and contractile function; 2) insulin sensitivity had been shown to be under circadian control in extracardiac tissues; and 3) our previous studies suggest that components downstream of PI3K in the insulin signaling cascade, namely, AKT and GSK3β, are regulated by the cardiomyocyte circadian clock (Abel, 2004; Durgan et al., 2010; Leighton et al., 1988). Quantitative RT-PCR confirmed that pik3r1 mRNA oscillates in control hearts in a time-of-day–dependent manner (with a periodicity of approximately 24 h) and that this oscillation is attenuated/abolished in CBK and CCM hearts (Fig. 4A; Suppl. Table S2). Decreased pik3r1 mRNA levels in CBK hearts does not appear to be due to expression of Cre alone, as α-MHC-Cre mouse hearts exhibit similar pik3r1 expression relative to wild-type littermates (1.00 ± 0.10 versus 1.06 ± 0.16 A.U. for α-MHC-Cre and wild-type littermate hearts isolated at ZT15, respectively; p > 0.05). Cosinor analysis revealed a significant (p < 0.05) circadian oscillation in pik3r1 mRNA levels in isolated cardiomyocytes following serum shock–induced synchronization (Fig. 4B).
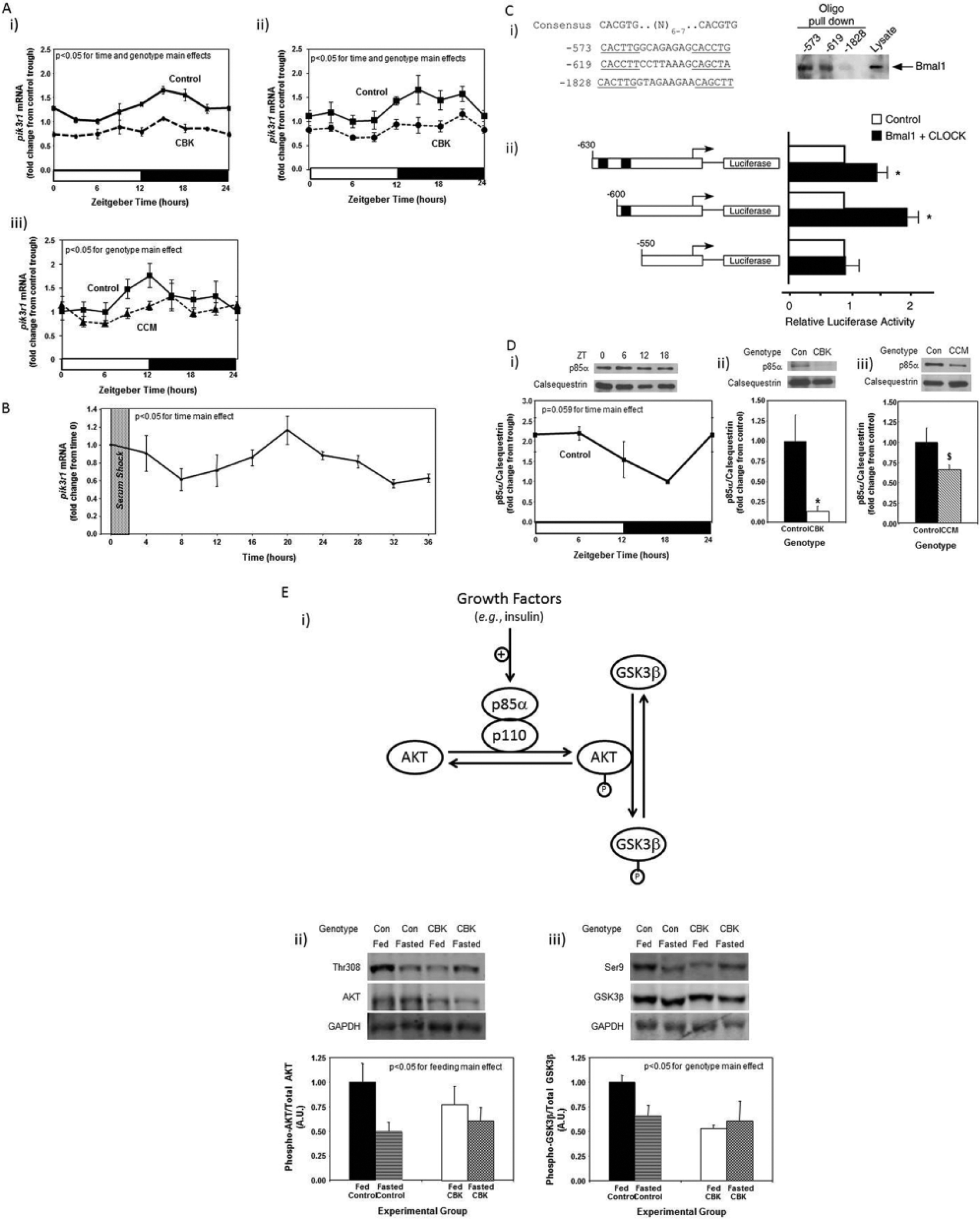
Alterations in time-of-day–dependent oscillations in pik3r1 mRNA in cardiomyocyte-specific Bmal1 knockout (CBK; microarray [i], RT-PCR [ii]) and Clock mutant (CCM; RT-PCR [iii]) hearts, compared with littermate controls (A). Persistence of circadian oscillations in pik3r1 mRNA in isolated adult rat cardiomyocytes following serum shock–induced synchronization (i.e., challenged with 50% serum for 2 h [shaded area]; B). Association of BMAL1 with two E-box consensus sequences in the Pik3R1 promoter (Ci); COS cells were transfected with expression vectors for Bmal1 and Clock, followed by DNA precipitation assay with biotinylated oligonucleotides (as described in the Materials and Methods section). BMAL1/CLOCK–mediated transcription of the Pik3r1 promoter (Cii); expression vectors for Bmal1 and Clock were co-transfected with Pik3r1 luciferase reporter and control plasmids into COS cells, as described in the Materials and Methods section (black boxes represent E-boxes at positions −619 and −573 in the Pik3r1 promoter). Diurnal variations in p85α protein levels in control hearts (Di). Decreased p85α protein levels in CBK (Dii) and CCM (Diii) hearts at ZT6. Relationship between PI3K, AKT, and GSK3β (Ei). CBK hearts exhibit phospho-Thr308-AKT (Eii) and phospho-Ser9-GSK3β (Eiii) levels that are comparable to fasted wild-type hearts. Mice were housed in a 12-h light:12-h dark cycle (lights on at ZT0). Hearts were isolated from 12-wk-old male CBK, CCM, and age-matched littermate controls. Note that ZT0 and ZT24 are identical data. Values are expressed as mean ± SEM (n = 3-13; sample size range varies depending on the parameter investigated). *p < 0.05 for CBK/CCM versus littermate control (post hoc pairwise comparison). $p = 0.076 for CCM versus control.
To investigate whether Bmal1 directly regulates Pik3r1, we examined the upstream regulatory sequence of the Pik3r1 promoter. We found several putative E-box sequences on the Pik3r1 promoter that could potentially recruit Bmal1. The putative E-boxes are located at −573, −619, and −1828 bp upstream of the Pik3r1 promoter. Notably, the putative E boxes located at −573 and −619 bp match very closely with the preferred BMAL1 binding motif, including their spacing and location immediately upstream of known Bmal1-regulated promoters (Rey et al., 2011). DNA precipitation assays using biotinylated oligonucleotides were performed to investigate whether BMAL1 binds directly to any of these 3 putative E-boxes. Figure 4Ci shows that BMAL1 was precipitated by the E-boxes located at −573 and −619 bp upstream of the Pik3r1 promoter. However, BMAL1 was not detected in the DNA precipitates using the E-box located at −1828 bp upstream of the Pik3r1 promoter. Next, we examined the function of the E-boxes located at −573 and −619 bp upstream of the Pik3r1 promoter using reporter gene assays (Fig. 4Cii). Co-transfection of BMAL1 and CLOCK increased Pik3r1 promoter activity. Deletion of E-boxes to −550 bp abrogated BMAL1/CLOCK–mediated increases in Pik3r1 promoter activity. Collectively, these observations suggest that Pik3r1 is directly regulated by the BMAL1/CLOCK heterodimer.
Pik3r1 encodes for p85α, the regulatory subunit of PI3K. Whether oscillations in pik3r1 mRNA observed in control hearts were associated with time-of-day–dependent fluctuations in p85α protein levels was next investigated; Figure 4D (and Suppl. Table S2) shows that p85α significantly oscillates in control hearts, peaking approximately 4 h into the light phase. In addition, p85α protein levels were decreased in both CBK and CCM hearts (90% and 34% decrease, respectively; Fig. 4D). We next investigated whether BMAL1-mediated regulation of Pik3r1 was associated with a functional impact on the downstream signaling components (illustrated in Fig. 4Ei), namely, AKT and GSK3β. Hearts were isolated during the dark phase (ZT20) from fed and 16-h fasted (fasted between ZT4 to ZT20) control and CBK mice. The purpose of investigating fed versus fasted mice was to compare basal versus insulin-stimulated states; data presented in Supplemental Figure S3 confirm higher plasma insulin levels in fed mice, independent of genotype. Consistent with the dramatic decreased in p85α levels in CBK hearts (90% lower compared with littermate controls; Fig. 4D), AKT phosphorylation at Thr-308 tended to be lower in CBK hearts (Fig. 4Eii). Importantly, phosphorylation of the AKT target, glycogen synthase kinase 3 beta (GSK3β), at Ser-9 was significantly reduced in hearts isolated from CBK mice (significant genotype main effect; Fig. 4Eiii); since the genotype-feeding status interaction term did not reach statistical significance (p = 0.06), pairwise comparisons between experimental groups was not possible. Collectively, our findings suggest that the cardiomyocyte circadian clock regulates both p85α and downstream signaling.
Physiologic Role of BMAL1 in the Adaptation of the Heart to Fasting
Based on our combined transcriptome and in silico analyses (summarized in Table 1) and subsequent validation studies (Figs. 3 and 4), it is clear that BMAL1 influences both myocardial metabolism (e.g., β-hydroxybutyrate oxidation) and signaling (e.g., the PI3K/AKT/GSK3β axis). To gain better insight regarding the extent to which BMAL1 influences these processes, we chose to perform a more in-depth analysis of substrate metabolism in CBK and littermate control hearts, in both the fed and fasted states. The scenario in which the animal in the wild is unsuccessful in foraging upon awakening was mimicked, by removing food from mice during their sleep time (light phase; ZT4), to ensure prevention of feeding upon awakening; all hearts were investigated at ZT20. Fasting influenced whole-body metabolism (e.g., respiratory exchange ratio [RER]) and humoral (e.g., plasma NEFA and insulin) factors in a similar fashion in CBK and littermate control mice (Suppl. Fig. S3). Independent of genotype, fasting increased fatty acid (oleate) oxidation, concomitant with decreased rates of glucose oxidation (i.e., p < 0.05 for feeding status main effects; Fig. 5A). Independent of feeding status, CBK hearts exhibit increased fatty acid (oleate) oxidation, concomitant with decreased rates of glucose oxidation, glycolysis (14C-lactate release), and net glycogen synthesis (i.e., p < 0.05 for genotype main effects; Fig. 5A). In addition, fasting decreased glucose oxidation in control, but not CBK, hearts (Fig. 5A). Finally, cardiac power was decreased in hearts isolated from fed (but not fasted) CBK mice; a similar trend was observed for a second contractile function parameter (i.e., rate pressure product; Fig. 5B). Collectively, these observations reveal a critical role of BMAL1 in myocardial substrate utilization.
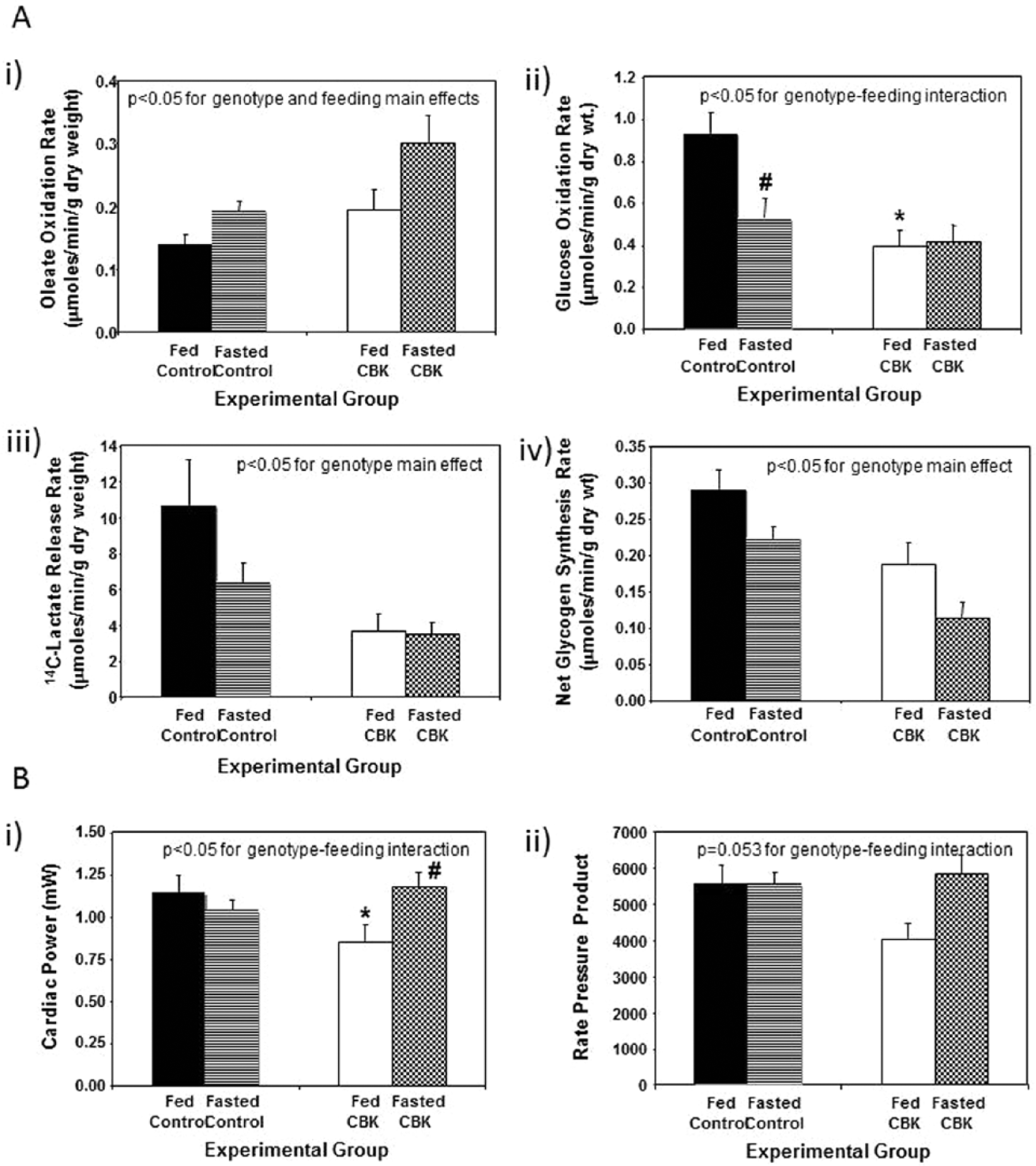
Response of cardiomyocyte-specific Bmal1 knockout (CBK) mice to a 16-h fast (relative to littermate control mice) at the level of substrate utilization (A) and contractile function (B). Mice were housed in a 12-h light:12-h dark cycle (lights on at ZT0); for fasted mice, food was removed from the cage at ZT4. All parameters represent values at ZT20. Values are expressed as mean ± SEM (n = 6-7; sample size range varies dependent on the parameter investigated). *p < 0.05 for CBK versus littermate control within a distinct feeding status (post hoc pairwise comparison). #p < 0.05 for fed versus fasted within a distinct genotype (post hoc pairwise comparison).
CBK Mice Exhibit Age-Onset Cardiomyopathy and Early Mortality
Maintenance of normal metabolic and signaling processes is critical for the heart. Given that these processes are aberrant in CBK hearts, we sought to investigate the long-term consequences of cardiomyocyte-specific BMAL1 deletion. Initially, serial echocardiography was performed in CBK versus littermate control mice (at 4-wk intervals, between 12 and 36 wk of age; Fig. 6). At 12 wk of age, echocardiographic parameters were not significantly different between CBK and littermate control mice (Fig. 6). By 20 wk of age, significant differences were observed with respect to genotype for systolic function parameters (fractional shortening and ejection fraction), which worsened with age (Fig. 6). Importantly, at 20 and 28 wk of age, α-MHC-Cre mice do not exhibit differences in systolic function parameters, relative to wild-type littermates (Suppl. Fig. S4), suggesting that, alone, the expression of Cre in cardiomyocytes is not sufficient to mediate the contractile dysfunction observed in CBK hearts.
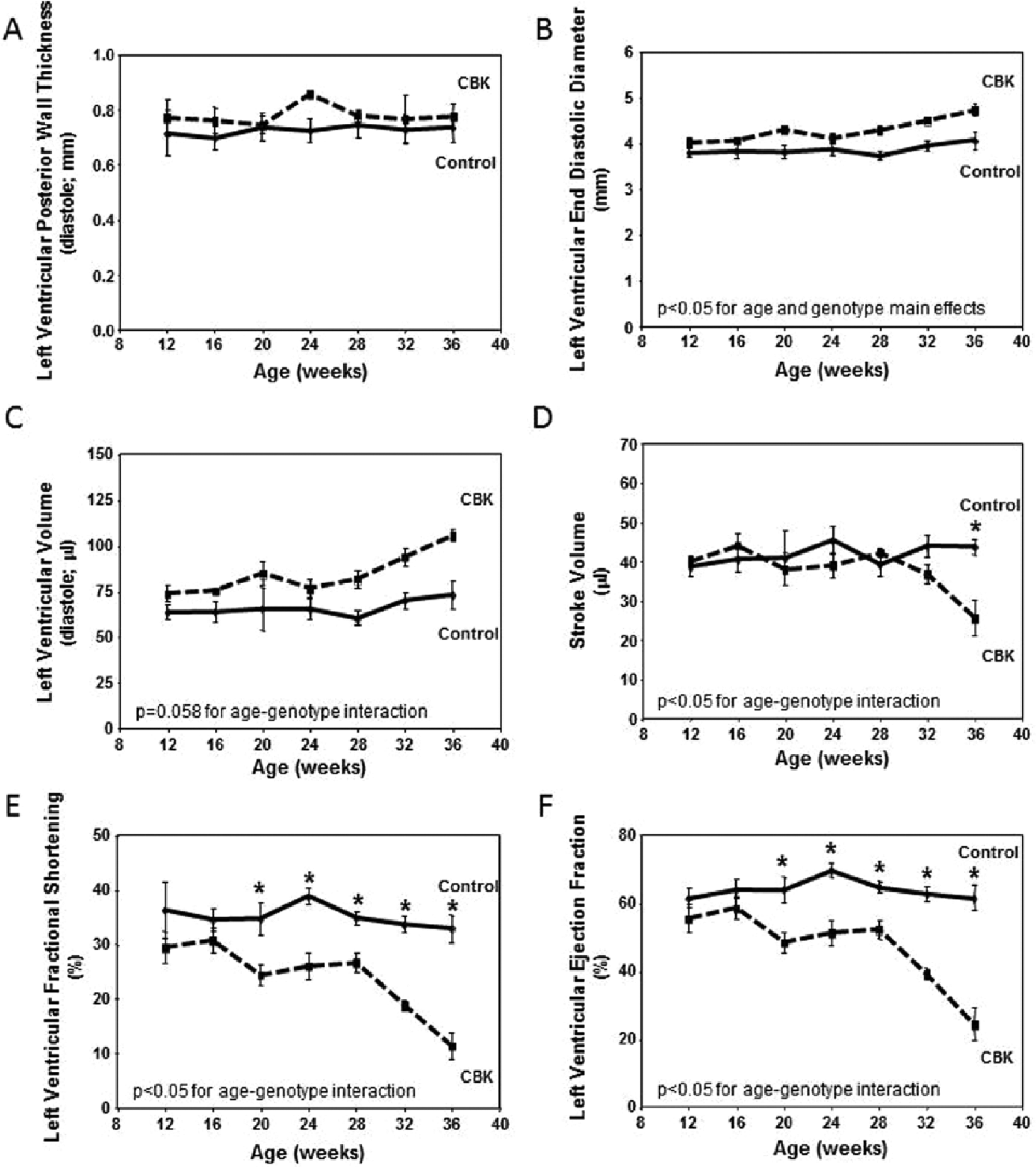
Age-dependent depression of cardiac function in cardiomyocyte-specific Bmal1 knockout (CBK), but not littermate control, mice. Left ventricular wall thickness during diastole (A), end diastolic diameter (B), left ventricular volume during diastole (C), stroke volume (D), left ventricular fractional shortening (E), and left ventricular ejection fraction (F). Mice were housed in a 12-h light:12-h dark cycle (lights on at ZT0). Serial echocardiography was performed at 4-wk intervals, always at ZT6. Values are expressed as mean ± SEM (n = 5-7). *p < 0.05 for CBK versus littermate control at a distinct age (post hoc pairwise comparison).
The age-dependent cardiomyopathy observed in CBK mice was next characterized at gravimetric, histological, and transcriptional levels. At 12 wk of age, no significant differences were observed between CBK and littermate control mice for any of the gravimetric parameters investigated (Table 2). In contrast, at 36 wk of age, biventricular weight, biventricular weight-to-body weight ratio, biventricular weight-to-tibia length ratio, and lung weight were all significantly elevated in CBK mice (relative to age-matched littermate controls; Table 2). Cardiomyocyte cross-sectional area was not significantly different between CBK and littermate control mice (age independent), although increased fibrosis was observed in hearts isolated from 36-wk-old CBK mice (Table 2). Collagen isoforms (i.e., col3a1 and col4a1 mRNA) and molecular markers of cardiac dysfunction (i.e., anf and mhcβ mRNA) investigated were significantly induced in hearts isolated from CBK mice at 36 wk of age, while serca2a mRNA was significantly repressed; cyclophilin mRNA served as a negative control (Table 2). Although col3a1 and col4a1 mRNA were slightly, but significantly, elevated in hearts isolated from CBK mice at 12 wk of age, molecular markers of cardiac dysfunction were not altered relative to littermate controls at this age (Table 2).
Associated with the development of age-onset cardiomyopathy, CBK mice exhibit a striking decrease in life span. Mean survival age of the CBK mice was 33 ± 3 wk versus 51 ± 0.7 wk in MHCα-Cre mice (p < 0.0001); no deaths were observed in either littermate control (i.e., BMAL1flox/flox) or wild-type mice during the 1-y study period (Fig. 7). Collectively, these data suggest that chronic genetic deletion of Bmal1 in cardiomyocytes results in cardiomyopathy and decreased life span.
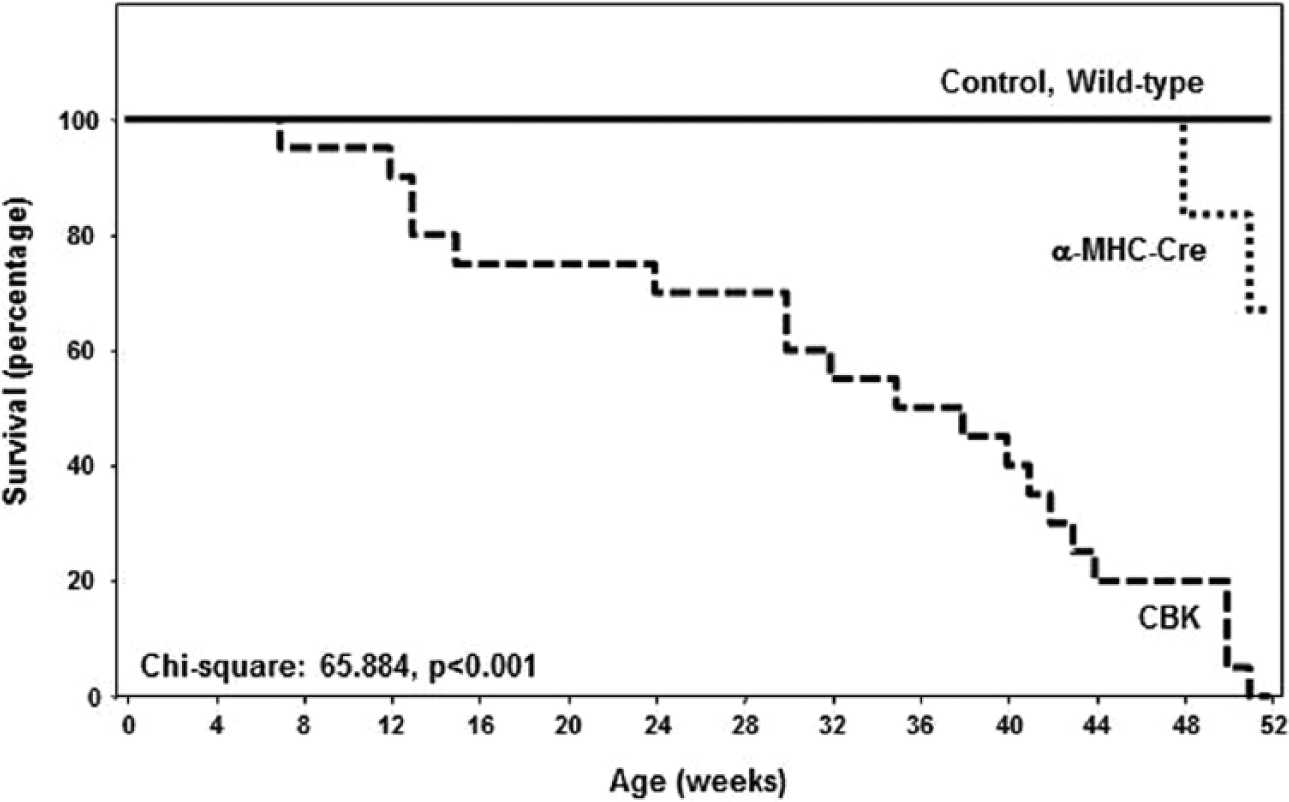
Decreased survival in cardiomyocyte-specific Bmal1 knockout mice compared with littermate controls. Values are expressed as mean ± SEM (n = 20).
Discussion
The purpose of the present study was to investigate the importance of BMAL1 in the heart, through the generation of CBK mice. Following validation of the cardiac-restricted nature of this model (Fig. 1; Suppl. Fig. S1), we identified 19 direct putative BMAL1 target genes, with known functions in metabolism and signaling (Table 1). Subsequent validation studies revealed that aberrant regulation of Bdh1 and Pik3r1 in CBK hearts was associated with impaired ketone body metabolism (Fig. 3) and signaling (Fig. 4), respectively. We next tested the hypothesis that a physiologic role of BMAL1 is for metabolic adaptation of the heart to fasting; we observed that CBK hearts exhibit a fasting-like metabolic profile (depressed glucose utilization and increased fatty acid oxidation), even in the fed state (Fig. 5). Finally, we report that chronic BMAL1 ablation in the heart results in an age-onset cardiomyopathy (Fig. 6) and reduced life span (Fig. 7; Table 2). Collectively, these studies highlight novel functions for the circadian clock component BMAL1 in the heart.
Animal models wherein circadian clock components are genetically ablated in a ubiquitous fashion have been used in attempts to reveal the relative contributions of this molecular mechanism on biology. These studies suggest that a plethora of critical processes, ranging from behavior (e.g., sleep-wake and feeding-fasting cycles), metabolic homeostasis (e.g., insulin secretion and responsiveness), and cellular function (e.g., proliferation), to longevity are under circadian clock control (Bass and Takahashi, 2010; Kondratov et al., 2006; Rudic et al., 2004; Takahashi et al., 2008; Turek et al., 2005). Two of the best well-characterized models, whole-body ClockΔ19 mutant and Bmal1 knockout mice, target the core heterodimer within the mammalian clock mechanism (Bunger et al., 2000; Vitaterna et al., 1994). However, despite the fact that CLOCK and BMAL1 function in the circadian clock mechanism in partnership, ClockΔ19 mutant and Bmal1 knockout mice do not always exhibit identical phenotypes. For example, ClockΔ19 mutant mice are obesity prone and exhibit normal life span, while Bmal1 knockout mice are lean and exhibit reduced life span (Bunger et al., 2005; Kondratov et al., 2006; Turek et al., 2005). Differences in phenotype have been explained in terms of the extent to which the circadian clock mechanism is affected (lesser residual circadian clock function in Bmal1 knockout mice) and/or functions of CLOCK and BMAL1 that are independent of the circadian clock mechanism. Appreciation of these reports underscores the need 1) to generate cell type–specific models to identify direct circadian clock functions in distinct organs and 2) to genetically manipulate multiple clock components, such that circadian clock–dependent and –independent mechanisms can be unveiled. Accordingly, we have generated 2 cardiomyocyte-specific models, namely, CCM and CBK mice (Durgan et al., 2006; Durgan et al., 2011b).
Multiple parameters of cardiac physiology and pathophysiology vary markedly as a function of time of day (Durgan and Young, 2010; Young, 2006). At a molecular level, previous microarray studies have revealed that approximately 10% to 15% of the cardiac transcriptome oscillates in a time-of-day–dependent fashion in wild-type mice (Martino et al., 2004; Storch et al., 2002). However, the relative contribution of factors extrinsic (e.g., neurohumoral factors) versus intrinsic (e.g., cell autonomous clock) to the heart is unknown. Through the use of CCM mice, we have previously reported that genetic disruption of CLOCK in the cardiomyocyte affects a host of genes whose protein products are involved in transcription, cellular signaling, ion homeostasis, and metabolism (Bray et al., 2008). With regard to the latter, we have revealed that wild-type hearts exhibit marked time-of-day–dependent rhythms in both glucose and fatty acid metabolism, which are dependent on cardiomyocyte CLOCK function (Bray et al., 2008; Durgan et al., 2011a; Tsai et al., 2010). The present study identified 19 putative direct BMAL1 target genes in the heart, several of which are involved in metabolism (Table 1). Consistent with identification of the triglyceride synthesis gene Dgat2 (Table 1), we have previously reported attenuated oscillations of this gene, and of triglyceride synthesis, in CCM hearts (Bray et al., 2008). Here, we uncovered a marked dependence of Bdh1 and ketone body metabolism on both CLOCK and BMAL1 (albeit in a time-of-day–independent manner; Fig. 3). Importantly, this is the first report suggesting that the circadian clock influences ketone body metabolism. Future studies are required to interrogate fully the links between the cell autonomous circadian clock and ketone body metabolism, as well as the potential implications during conditions of increased ketone body utilization (e.g., starvation, diabetes).
Several putative direct BMAL1 target genes identified in our analysis are known to play a role in cellular signaling (Table 1). These genes are of particular interest, as they might provide insight with regard to which extracellular stimuli/stresses the cardiomyocyte circadian clock allows the heart to anticipate on a daily basis. One such gene was Pik3r1, which encodes for an insulin signaling component (i.e., the p85α subunit of PI3K). We have previously reported that the phosphorylation status of 2 kinases that are downstream of PI3K (namely, AKT and GSK3β) appear to be under circadian clock control (as evidenced by loss of time-of-day–dependent oscillations in CCM hearts; Durgan et al., 2010; Ko et al., 2011); similar findings regarding clock regulation of AKT phosphorylation have been reported in extracardiac tissues, using germline Bmal1 null and Per2 mutant mice (Anea et al., 2009; Carvas et al., 2012). Consistent with impaired PI3K activity, both AKT and GSK3β phosphorylation were attenuated in CBK hearts (Fig. 4E). Collectively, these data are consistent with direct regulation of Pik3r1 and downstream signaling by the cardiomyocyte circadian clock. This concept is supported by previously published studies, reporting that diurnal variations in whole-body insulin sensitivity are influenced by circadian clocks (Rudic et al., 2004). Importantly, the current study is among the first to highlight a potential mechanism by which cell autonomous circadian clocks directly affect the PI3K/AKT/GSK3β signaling axis.
A primary goal of the current study was to determine 1 (or more) physiologic role(s) for BMAL1 in the heart. Given that BMAL1 directly influenced genes involved in metabolism (e.g., ketone body metabolism) and signaling (e.g., insulin signaling), we hypothesized that BMAL1 may play a role in the metabolic adaptation of the heart to fluctuations in feeding status. Consistent with this concept, previous studies have highlighted a role for BMAL1 and/or the circadian clock in both whole-body and hepatic responsiveness to fasting (Lamia et al., 2008; Zhang et al., 2010). In addition, we have previously observed a differential transcriptional response of CCM hearts to a 12-h fast (Durgan et al., 2006). In an attempt to mimic the scenario in which the mouse in the wild is unsuccessful in foraging upon awakening, food was removed from mice during the sleep (light) phase (thereby preventing food consumption upon awakening). We observed that even in the fed state, CBK hearts exhibit a fasting-like metabolic phenotype (increased fatty acid oxidation and decreased glucose utilization), which is consistent with the metabolic profile of CCM hearts (Bray et al., 2008; Durgan et al., 2011a). We speculate that depressed PI3K/AKT/GSK3β signaling in CBK hearts (Fig. 4E) signals a chronic fasted state. It should be noted that these observations are somewhat in contrast with recent reports that fatty acid oxidation is depressed in isolated mitochondria from germline Bmal1 null mouse livers (Peek et al., 2013). However, we speculate that investigation of metabolism in an intact organ likely preserves extra-mitochondrial regulatory points, including substrate supply (e.g., control of fatty acyl-CoA uptake into the mitochondrial matrix is a critical β-oxidation regulatory mechanism in intact tissues/cells, which is abolished in isolated mitochondrial studies). Collectively, these observations confirm an important role for BMAL1 in the regulation of myocardial metabolism, as well as in the adaptation of the heart to fasting (i.e., a physiologic stress).
Given that BMAL1 influences a number of important processes in the heart (e.g., metabolism and signaling), we sought to investigate the consequence of chronic genetic ablation of this transcription factor in a cardiomyocyte-specific manner. We have previously reported that both CCM and CBK hearts exhibit a prohypertrophic phenotype (Durgan et al., 2011b). Consistent with these observations, the current study reports that CBK mice develop an age-dependent cardiomyopathy (Fig. 6), which is associated with a decreased life span (Fig. 7). This phenotype is remarkably similar to that reported for germline BMAL1 null mice, wherein systolic dysfunction begins at 20 to 24 wk of age (Lefta et al., 2012).
It is important to acknowledge a number of shortcomings and unanswered questions associated with the current study. For example, identification of the precise mechanism(s) by which loss of BMAL1 in the heart results in cardiomyopathy has not been determined. Our analyses clearly reveal that BMAL1 influences a number of critical functions in the heart, leading to the suggestion that the cardiomyopathy observed in CBK mice is potentially multifactorial, as opposed to disruption of a single physiologic process. It is highly likely that the conservative nature of our in silico analysis resulted in a large number of false-negatives, in terms of identification of putative BMAL1 target genes. One contributing factor was use of liver ChIP sequencing data to guide the analysis; this may explain why genes such as Klf15 and Scn5a, which have previously been suggested to be regulated by Bmal1 in the heart (Jeyaraj et al., 2012; Schroder et al., 2013), were not included in Table 1. It should also be noted that all animal studies were performed under light/dark conditions, raising the possibility that a subset of genes oscillate in the heart secondarily to light stimulation. Finally, the reason why a subset of BMAL1- and CLOCK-regulated genes (such as Bdh1) do not oscillate in the heart is also not known; it is possible that this is due to a long half-life for these mRNA species.
In summary, the current study has revealed novel functions for BMAL1 in the heart, including regulation of substrate utilization (e.g., ketone body, fatty acid, and glucose metabolism) and signaling (e.g., the PI3K/AKT/GSK3β signaling axis). In addition, a functional consequence of chronic genetic ablation of BMAL1 in the cardiomyocyte is development of age-onset cardiomyopathy and reduced life span. These studies therefore highlight critical functions for BMAL1 in the heart.
Footnotes
Acknowledgements
This work was supported by the National Heart, Lung, and Blood Institute (HL-074259, HL-106199, and HL-107709). Bradley W. Peden was supported by an American Heart Association Fellowship (MSRF9960000). Billy-Joe Ammons was supported by the University of Alabama at Birmingham Summer in Biomedical Sciences (SIBS) Undergraduate Research Program. Analysis of microarray data was performed at the UAB Heflin Center for Genomic Sciences. Echocardiography was performed by the UAB Diabetes Research Center Small Animal Physiology Core (DK11015). Humoral factor analysis was performed by the UAB Metabolism Core Laboratory for the Nutrition Obesity Research Center (P30 DK56336, UL 1RR025777, P60DK079626). We would like to thank Alan Akira, Maximiliano H. Grenett, Betty M. Pat, and William F. Ratcliffe for technical assistance.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.