
Abstract
An internal circadian clock regulates the electrical activity of cardiac myocytes controlling the expression of potassium channel interacting protein–2 (KChIP2), which is a key regulator of cardiac electrical activity. Here, we examine how the circadian rhythm of KChIP2 expression affects the dynamics of human and murine ventricular action potentials (APs), as well as the intervals in the equivalent electrocardiograms (ECGs) reflecting the duration of depolarization and repolarization phases of the cardiac ventricular APs (QRS and QT intervals), with mathematical modeling. We show how the internal circadian clock can control the shape of APs and, in particular, predict AP, QRS, and QT interval prolongation following KChIP2 downregulation, as well as shortening of AP, QRS, and QT interval duration following KChIP2 upregulation. Based on the circadian expression of KChIP2, we can accurately predict the circadian rhythm in cardiac electrical activity and suggest the transient outward potassium currents as the key current for circadian rhythmicity. Our modeling work predicts a smaller effect of KChIP2 on AP and QT interval dynamics in humans. Taken together, these results support the role of KChIP2 as the key regulator of circadian rhythms in the electrical activity of the heart; we provide computational models that can be used to explore circadian rhythms in cardiac electrophysiology and susceptibility to arrhythmia.
Circadian rhythms in cardiac electrical activity have been observed in humans and a variety of domestic mammals (Piccione et al., 2005). These circadian rhythms are likely created by altering the expression of ionic currents in the heart, which lead to changes in the morphology and dynamics of cardiac action potentials (APs), such as prolongation or attenuation of the AP as well as QT interval durations, and slower conduction velocity. Changing these factors can increase the susceptibility toward the development of reentrant arrhythmias (Antzelevitch et al., 2007; Cheng et al., 2011; Jeyaraj et al., 2012; Kuo et al., 2001), perhaps explaining why susceptibility of the occurrence of ventricular arrhythmias peaks during the morning hours (Guo and Stein, 2003; Portaluppi and Hermida, 2007). Potassium channel interacting protein–2 (KChIP2) expression, a regulator of cardiac ionic conductance, as well as the QT interval durations, shows circadian rhythmicity (Jeyaraj et al., 2012), suggesting its role in regulation of cardiac circadian rhythms. Furthermore, it has been indicated that absence and excess of KChIP2 are directly correlated to abnormal repolarization, nonrhythmic QT interval duration, and, hence, higher susceptibility to the development of ventricular arrhythmias in mice (Jeyaraj et al., 2012). Thus, understanding how circadian rhythms in cardiac electrophysiology are established, and the role of KChIP2 in generating these rhythms, remains an important scientific goal.
Perhaps the most established use of mathematical modeling in biology is to study how the complex coordinated action of the many cardiac ionic currents regulates the electrical activity of the heart (Pandit et al., 2001; Grandi et al., 2010). Here we consider 2 mathematical models of cardiac electrophysiology. One models human ventricular myocytes and is based on a model published by Grandi et al. (2010), and the other models mouse ventricular myocytes and is based on a modified Pandit et al. (2007) model (further information about these models is provided in the Methods section of the supplementary online material). In addition, the mammalian circadian clock has been studied using modeling (Sim and Forger, 2007; Belle et al., 2009; Dean et al., 2009; Ko et al., 2010). This has motivated us to study how the circadian clock affects the electrical function of the heart, and specifically the AP dynamics of human and murine ventricular myocytes, using mathematical models.
No mathematical model has yet been used to study cardiac circadian rhythms. We modify a murine mathematical model and human mathematical model of cardiac electrical activity to account for the circadian rhythm of KChIP2 and its effects (see the Methods section and the supplementary online material). The behavior of our computational model matches data from a recent study by Jeyaraj et al. (2012) on KChIP2 and makes predictions about the important ion channels in circadian regulation of cardiac activity and the differences between circadian regulation in the murine and human heart.
Introduction to Cardiac Action Potentials and the Role of KChIP2
In a cardiac AP, the cardiac sodium current (INa) is responsible for the rapid depolarization phase by providing a rapid influx of sodium ions in the cell. When the Na+ channels start inactivating, a net outward flow of potassium and chloride ions follows, undertaken primarily by the calcium-independent transient outward potassium current (Ito). This contributes to the formation of the “notch” observed in the AP. The observed “plateau” phase of the AP is maintained by an influx of Ca2+ ions through the L-type calcium current (ICa,L) and an outflow of K+ ions through the slow component of the delayed rectifier potassium current (IKs). The Na+/Ca2+ exchanger current (Incx), the Na+/Ca2+ pump current (INaK), and some background Na+ and Ca2+ currents (INabk and ICabk) partially contribute at the same time. Moreover, the repolarization phase of the AP is characterized by an outflow of potassium ions through the rapid component of the delayed rectifier potassium current (IKr) and IKs, closing of ICa,L, and activation of the inward rectifier K+ current (IK1), with the latter playing an essential role in the resetting of the membrane potential back to the resting potential.
Even though the ionic currents that contribute to the formation of the AP are highly conserved between the 2 species studied here (human versus murine), there are electrophysiological differences. First, the duration of the human AP is longer than that of the murine AP, which has been attributed to the higher heart rates (~10 times) of mice (Salama and London, 2007). Also, the major repolarizing currents are different between the 2 species. In humans, the major repolarizing currents are IKs and IKr, whereas in mice, repolarization is governed by Ito as well as IKs (Fermini and Fossa, 2003; Salama and London, 2007).
To effectively study the cardiac action potential, the QT and QRS intervals need to be examined. In an ECG, the QT interval represents the duration and shape of the depolarization and repolarization phases of the cardiac ventricular AP, whereas the QRS complex depicts only the depolarization phase of the cardiac ventricular AP. Usually, due to the dependence of the QT interval in heart rate, a corrected QT (QTc) interval is calculated, with the most common way being through Bazett’s formula (Bazett, 1920; Goldenberg et al., 2006). In mice, typical QTc interval and QRS complex durations are around 54 msec and 22 msec, respectively (Mitchell et al., 1998; Wehrens et al., 2000), whereas in humans, typical QTc interval and QRS complex durations are in the range of 360-440 msec and 80-90 msec, respectively (Goldenberg et al., 2006; Shimizu et al., 2000).
The KChIPs were originally described as a group of 4 alternatively spliced auxiliary subunits for the rapidly inactivating (A-type) voltage-gated potassium (Kv) currents in neuronal and cardiac tissue (An et al., 2000; Thomsen et al., 2009b). They belong to the family of neuronal calcium sensor proteins and contain 4 EF-hand motifs, of which only the last 3 bind Ca2+ (Thomsen et al., 2009b). Of all KChIPs, KChIP2 is the only one preferentially expressed in the heart. It regulates the fast component of Ito (Ito,fast) by selectively interacting with the N-termini of Kv4 channels and most notably Kv4.2 and Kv4.3 (An et al., 2000; Kuo et al., 2001; Rosati et al., 2001, 2003). Additionally, KChIP2 has been found to regulate several other cardiac ion currents (Thomsen et al., 2009b): Ito,slow (Deschênes et al., 2008), ICa,L (Thomsen et al., 2009a, 2009b), INa (Deschênes et al., 2008; Meadows and Isom 2005), and IKs (Li et al., 2005; Cheng et al., 2011). Downregulation of KChIP2 leads to decreased expression of Ito,fast (Kuo et al., 2001; Patel et al., 2004; Shibata et al., 2003), reduced ICaL density (Thomsen et al., 2009a, 2009b), increased expression of IKs (Cheng et al., 2011), and reduced Ito,slow and INa density expression (Deschênes et al., 2008). Moreover, upregulation of KChIP2 causes increased expression of Ito,fast (Jin et al., 2010) as well as increased ICaL channel density (Thomsen et al., 2009b).
Methods
Construction of the Human and Murine Computational Models
To construct the 2 computational models (human and murine) describing the dynamics of KChIP2 and how it affects the behavior of certain cardiac ion channels, we altered 2 previously published models simulating the electrophysiology of human and murine ventricular myocytes, respectively. Specifically, the human model used describes the electrophysiology of human ventricular myocytes based on the experimental data reported by Grandi et al. (2010). The murine model used describes the electrophysiology of murine ventricular myocytes and was based on a modified version of a published model by Pandit et al. (2001) (see the Methods section of the supplementary online material).
The main method followed in order to create the models was as follows: Suppose X is the current affected by KChIP2. As discussed above, X can be Ito, ICaL, INa, and IKs. Generally, currents in our models are of the following form:
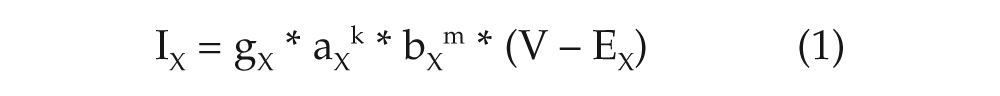
where gX is the maximal conductance of X, a is an activation gating variable raised to a power k depending on the model, b a deactivation gating variable raised on the mth power (depending on the model), V the membrane potential, and EX the resting potential of X. Since KChIP2 affects the dynamics of that current, it needs to be included in the above equation. Let M be a function of the concentration of KChIP2 describing the rate at which the current density of X is altered. It is assumed that M is described by Michaelis-Menten kinetics in the currents ICaL, INa, and IKs, and with a normalized linear relationship for Ito (this is equivalent to using a Michaelis-Menten relationship with a relatively low K). The reason Ito is treated differently is because it has been reported that when KChIP2 is upregulated by a factor of 2, the density of Ito increases by a factor of 2 as well (Jin et al., 2010; Jeyaraj et al., 2012).
Thus, in our model, for X equal to ICaL, INa, and IKs:
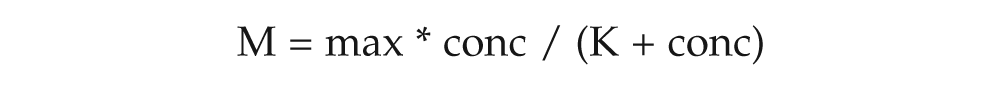
and for X equal to Ito:

where max is the maximum rate achieved by the system at saturating protein concentrations, K is the concentration of KChIP2 when the rate is equal to half of max, and conc is the concentration of KChIP2. Consider the case where KChIP2 is absent. In that case, according to the above equations, M should be 0. However, in some reported cases, some currents still persist in the absence of KChIP2. For this reason, a new term needs to be introduced. Let rateinitial be the rate at which the current density of X is altered when KChIP2 is absent. This term is determined by experimental data depending on the currents examined. Specifically, rateinitial was set equal to the average of ratios of current density of X in the presence of KChIP2 over X alone, experimentally found in the publications mentioned at the beginning of this article, over different voltages. The actual rate constants we used are provided in Supplementary Table S1.
Implementing those new terms in Equation 1, an updated equation describing the current density of X that depends on KChIP2 dynamics is presented:
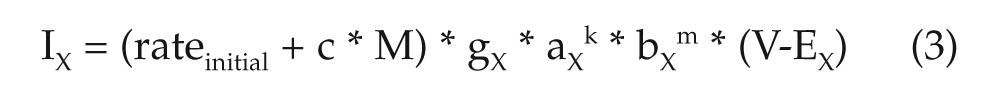
When the concentration of KChIP2 is normal, the term (rateinitial + c * M) should be exactly equal to 1, in order to match the previously published models. For this reason, max was set equal to (1 – rateinitial) (note that during physiological conditions, M is near saturation), and c was set equal to the ratio of (K + normal_concentration_KChIP2) over normal_concentration_KChIP2 for ICaL, IKs, and INa and equal to 1 for Ito.
In the human computational model, the densities of the ICaL, INa, and IKs currents are each expressed as a combined result of a subsarcolemmal and a junctional component. In our human computational model, the method described above (and hence Equation 3) was used only to describe the subsarcolemmal compartment of each current. This is due to the fact that the experimental data used to construct the models were reporting the behavior of cardiac ion channels (under normal, absent, and upregulated KChIP2 concentrations) on the cell membrane and not that much in the junctional compartment of the cardiomyocytes. Also, the Ito of the human model distinguishes a fast and a slow component (Ito = Ito,fast + Ito,slow). Therefore, in our human computational model, the rateinitial and M parameters of those 2 components differ from each other (specific numbers and sources are provided in Suppl. Table S1).
Modeling the Concentration of KChIP2
To account for the potential circadian variation patterns in KChIP2 expression examined in the introduction, the concentration of KChIP2 was set to be sinusoidal. Specifically:
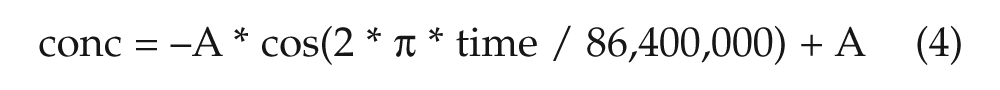
where A is a constant depending on the average concentration of KChIP2. Without loss of generality, it is assumed throughout this paper that A = 200 represents relatively normal KChIP2 concentration. The period is set equal to 86,400,000 (msec) to account for the circadian variation of 24 h. Moreover, the cosine function was chosen in order for the KChIP2 expression to peak at CT12 and show a trough at CT0, for the model to accede with the experimental predictions of Jeyaraj et al. (2012). Generally, when KChIP2 is absent, conc is set equal to 0; when KChIP2 is at relatively normal concentration, conc is set equal to 200; and when KChIP2 is upregulated, conc is set equal to 400. Details on our parameter choices and simulation methods are provided in the Methods section of the supplementary online material.
Results
In Figure 1, the results of our murine computational model are compared with the results recently published by Jeyaraj et al. (2012) describing the behavior of KChIP2 in murine ventricular myocytes. Both our computational murine model and the experimental data reported by Jeyaraj et al. (2012) predict prolonged AP, QRS, and QT interval durations when KChIP2 is knocked out (Fig. 1A, 1B, 2C). When upregulated, there is a distinct shortening of the AP as well as the QRS and QT interval durations (Fig. 1A, 1B, 2C). The upstroke velocity of the APs in the computational model is less when KChIP2 is absent and more when KChIP2 is upregulated, compared with the relatively normal case (Fig. 1A). Moreover, the QRS and QT interval durations in both the computational model and the experimental murine data are highest when KChIP2 is absent and lowest when it is upregulated (Fig. 1A, 1B, 2C).
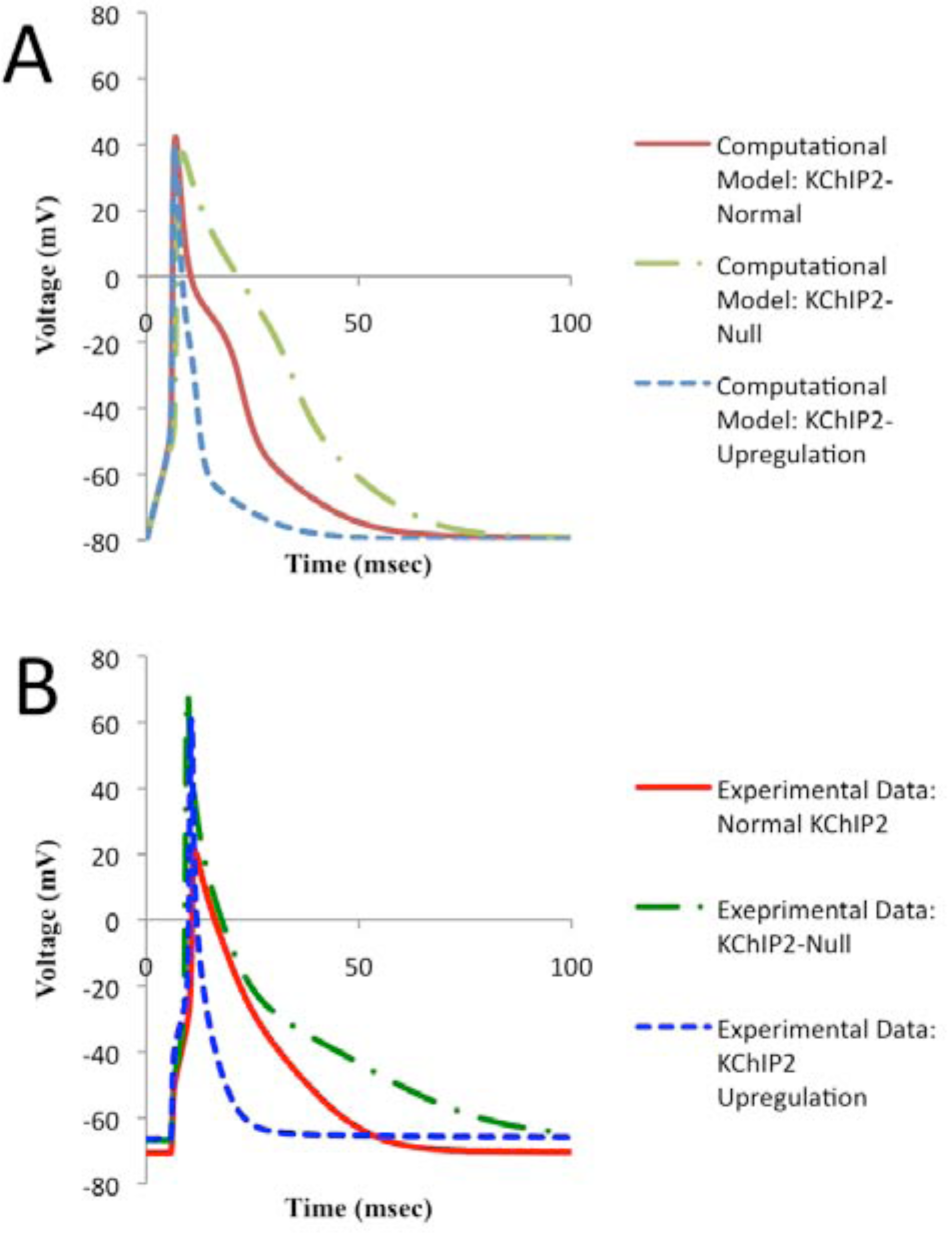
APs based on our murine computational model and compared with experimental data by Jeyaraj et al. (2012). (A) APs predicted by our model in response to relatively normal concentrations, downregulation, and upregulation of KChIP2, a key circadian regulator of cardiac electrophysiology. (B) APs corresponding to relatively normal concentrations, absence, and upregulation of KChIP2 from the experimental data published by Jeyaraj et al. (2012)
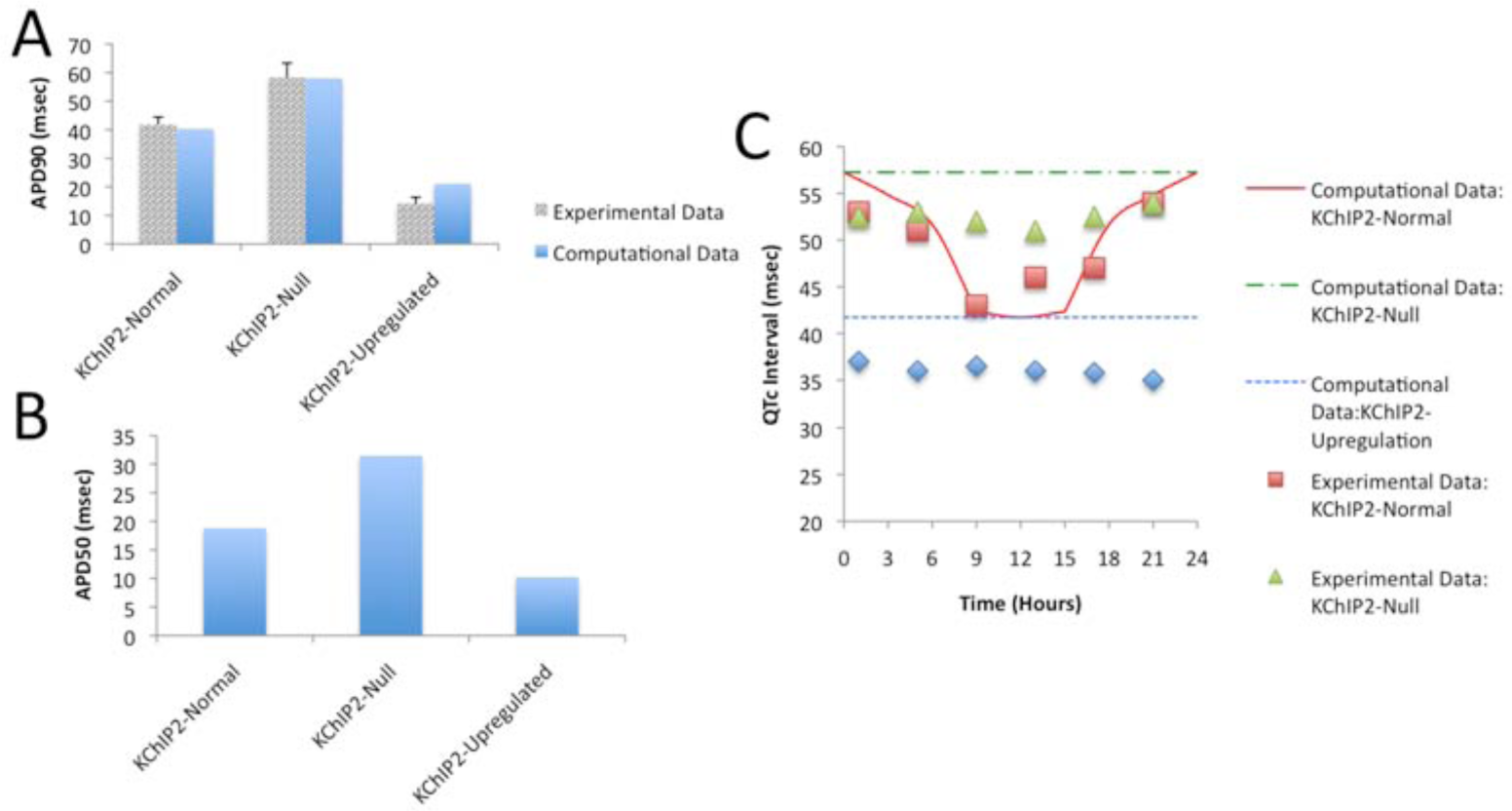
APD90s, APD50s, and QTc interval durations based on our murine computational model and compared with experimental data by Jeyaraj et al. (2012). (A) APD measured at 90% of repolarization under relatively normal, downregulated, and upregulated KChIP2 concentrations. The APD90s occurring from our proposed murine computational model are compared with experimental data from Jeyaraj et al. (2012). (B) APD measured at 50% of repolarization occurring from our murine computational model under relatively normal, downregulated, and upregulated KChIP2 concentrations. (C) QTc interval duration over a 24-h period. The QTc intervals occurring from our proposed murine computational model are plotted against the QTc intervals experimentally reported by Jeyaraj et al. (2012). As in Parts A and B, the results reflect relatively normal, downregulated, and upregulated concentrations of KChIP2. Taken together, these results show that our computational model is accurate in predicting the effect of KChIP2 and corresponding circadian variation in murine ventricular myocyte electrophysiology.
The main difference between the experimental data and our computational model is in the peaks of the APs. The experimentally reported peaks are located between 20 mV and 70 mV, whereas in our computational models, the peaks of the APs are all around 50 mV (Fig. 1). In addition, in the reported experimental data (Jeyaraj et al., 2012), under relatively normal concentrations of KChIP2, the resting membrane potential is more negative than under the null and the upregulated cases (Fig. 1B), whereas in our computational model the resting membrane potentials are the same under all 3 cases (Fig. 1A). These differences do not affect our overall results and may be caused by cell-to-cell variability in experiments.
The APD90s for both the computational model and experimental data match well (Jeyaraj et al., 2012) (Fig. 2A). Our methods for calculating the AP and QT interval durations are provided in a dedicated section of the supplemental online material. Specifically, the overall changes in the computational versus experimental APD90s were approximately 37 and 44 msec, respectively (Fig. 2A). We additionally measured the APD50s for the computational model, and the overall change was approximately 21 msec (Fig. 2B). Moreover, the model faithfully reproduces the circadian variation in QT interval duration observed by Jeyaraj et al. (2012) with a minimum at CT12 and maximum at CT0 and predicts that when KChIP2 is absent or upregulated, no distinct oscillatory expression is observed since circadian effects in our model are mediated by KChIP2 (Fig. 2C).
To understand how KChIP2 contributes to the shape of the occurring APs of our murine computational model, we examined each of the currents that KChIP2 controls by removing the KChIP2 regulation (and thus setting to normal rates) from all currents except for the one being studied. KChIP2’s regulation of Ito has the most pivotal effect on the shape of the AP and especially on the repolarization phase of the cardiac AP (Fig. 3A). Knocking out the KChIP2 regulating Ito increases the APD by approximately 50 msec, whereas upregulating it decreases the APD by approximately 20 msec (Fig. 3A). The effect that the other currents have on the shape of the occurring AP is less strong than that of Ito but nonetheless important (Fig. 3, B-D). Specifically, as seen in Figure 3B, knocking out the KChIP2 regulating ICaL causes the AP plateau (Phase 2) to significantly diminish, which decreases the APD. On the contrary, upregulating KChIP2 increases slightly the duration of Phase 2 but has no significant effect on the APD (Fig. 3B). Affecting the KChIP2 regulating IKs has a similar effect as ICaL, and altering the KChIP2 modulating INa causes the APD to increase slightly when KChIP2 is knocked out and has no significant effect when KChIP2 is upregulated (Fig. 3, C and D). Generally, even though Ito plays the most significant role in determining the shape of the occurring APs, the other currents are essential to balance out the effect of Ito.
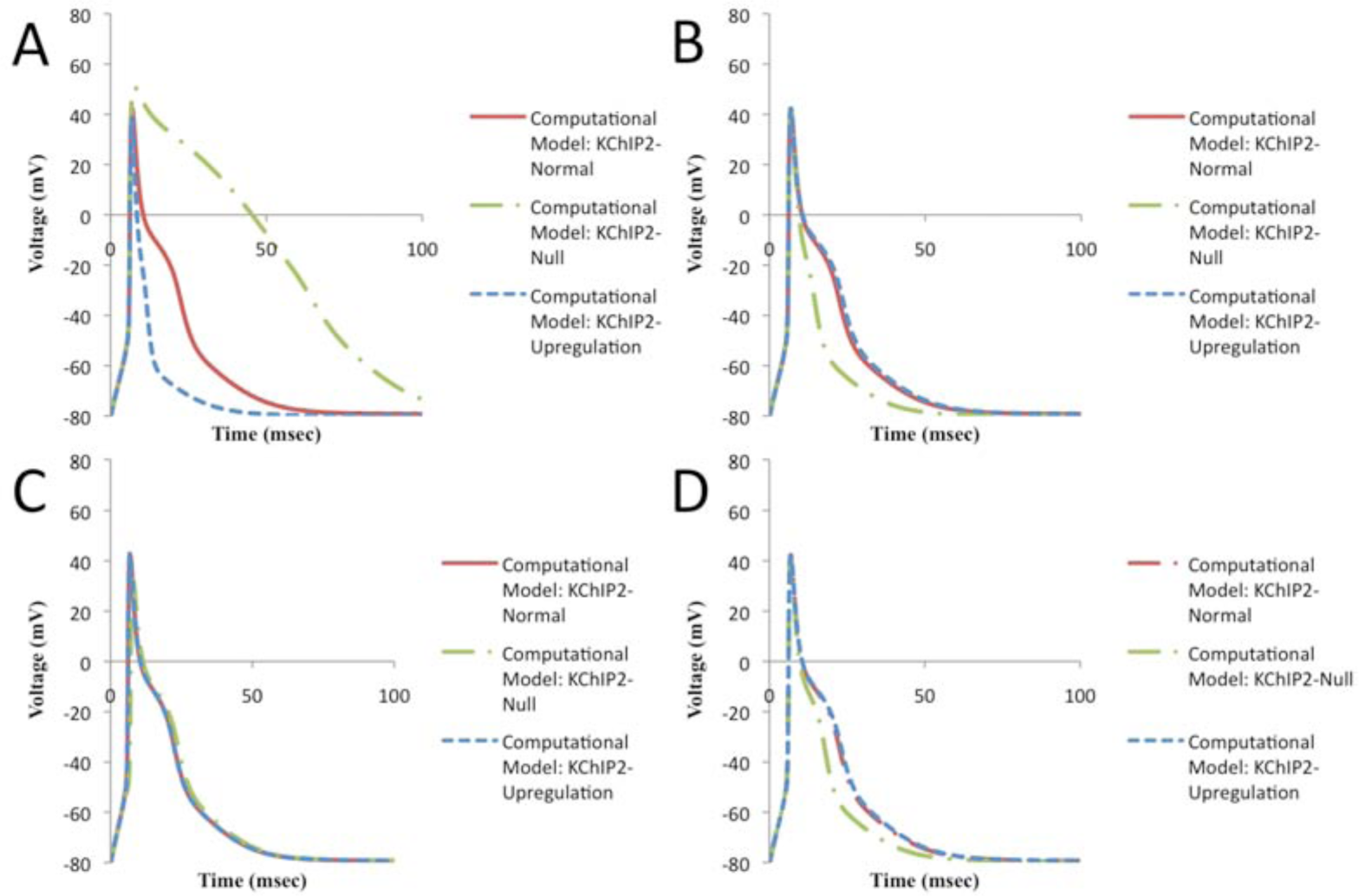
Model predictions on how each current regulated by KChIP2 contributes to the shape of the murine ventricular AP. (A) Ito: Resulting APs when the KChIP2 regulating only Ito is set to relatively normal, downregulated, and upregulated concentrations. (B) ICaL: Resulting APs when the KChIP2 regulating only ICaL is set to relatively normal, downregulated, and upregulated concentrations. (C) INa: Resulting APs when the KChIP2 regulating only INa is set to relatively normal, downregulated, and upregulated concentrations. (D) IKs: Resulting APs when the KChIP2 regulating only IKs is set to relatively normal, downregulated, and upregulated concentrations
Overall, our murine computational model is able to reproduce with great accuracy the experimentally observed behavior and dynamics of KChIP2 on murine ventricular myocytes. This not only argues for our model being an important potential tool in understanding the circadian variation in cardiac electrical activity but also shows how the circadian variation in KChIP2 expression, proposed by Jeyaraj et al. (2012), is consistent in explaining the circadian variation of murine ventricular myocyte AP shape.
To predict how this circadian variation in KChIP2 might affect human ventricular myocyte AP dynamics, we added our KChIP2 model to a mathematical model of human ventricular myocyte electrical activity. The resulting APs are shown (Fig. 4). Prolongation and shortage of the APDs (APD50 and APD90) are noticed when KChIP2 is absent and upregulated, respectively (Fig. 5, A and B). Specifically, the total change observed in APD50s is approximately 17 msec, whereas the total change observed in APD90s is approximately 23 msec (Fig. 5, A and B). In addition, the QRS and QT intervals are accentuated, and a significant J-wave elevation is discerned when KChIP2 is absent (Fig. 4, 5C). The latter causes elevation of the ST segment on the equivalent ECG. The elevation of the J-wave is due to the suppression of the Ito, which in humans is responsible for the formation of the notch of the AP. This is the reason why when KChIP2 is absent, the notch is eliminated (Fig. 4), matching experimental data by Kuo et al. (2001) and Thomsen et al. (2009a).
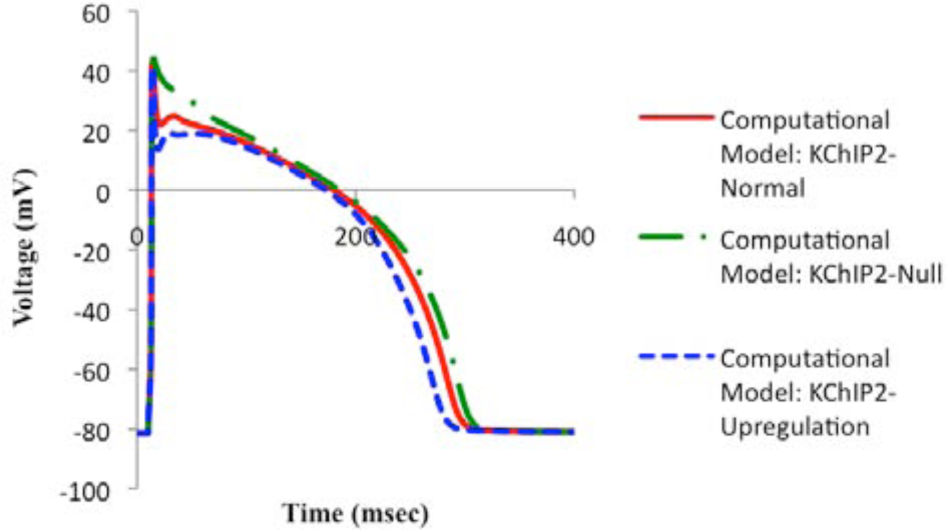
APs based on our human computational model under relatively normal, downregulated, and upregulated concentrations of KChIP2.
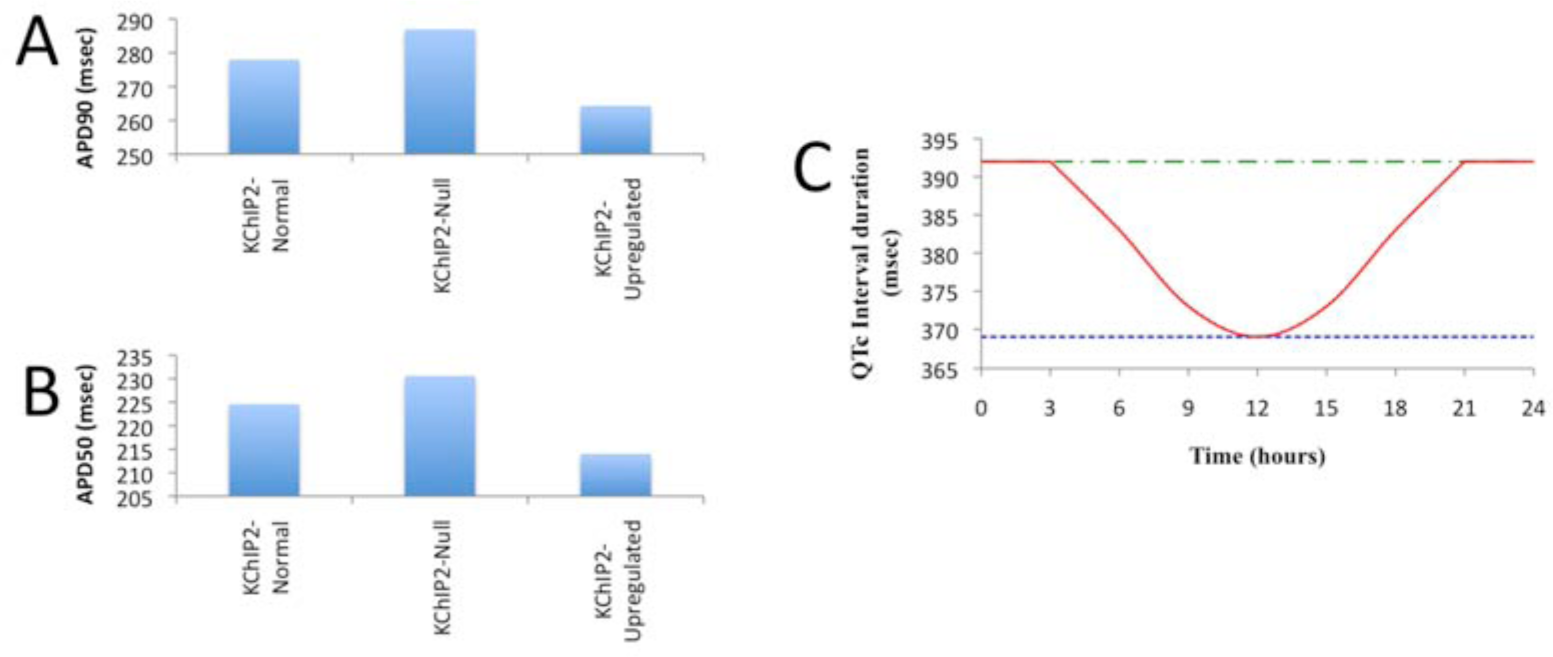
Model predictions of the effects of KChIP2 and circadian variation in human ventricular myocyte electrophysiology. (A) APD90s, (B) APD50s, and (C) QTc interval durations constructed similarly as in Figure 2. This model predicts similar but attenuated effects of KChIP2 in humans compared with mice.
The QT interval duration over a 24-h period presents the same trend as in the murine model, that is, oscillatory behavior under relatively normal concentration of KChIP2 and overall stable duration under absence and upregulation of KChIP2 (Fig. 5C).
Similarly, the effect of each current regulated by KChIP2 on the shape of the AP was examined. Of all currents, Ito,fast is shown to have the biggest effect on the shape of the AP and especially on the repolarization phase (Fig. 6A). Moreover, ICaL and IKs result in somewhat significant changes in the APD when KChIP2 is knocked out (Fig. 6, C and E). The other currents do not seem to produce significant changes in the shape of AP when the concentration of the KChIP2 affecting them individually is altered (Fig. 6, B and D). According to our human computational model, even though Ito,fast seems to play the most important role in the shape of the AP, the other currents, and especially ICaL and IKs, play an important role in balancing the effect of Ito,fast in the morphology of the human ventricular AP.
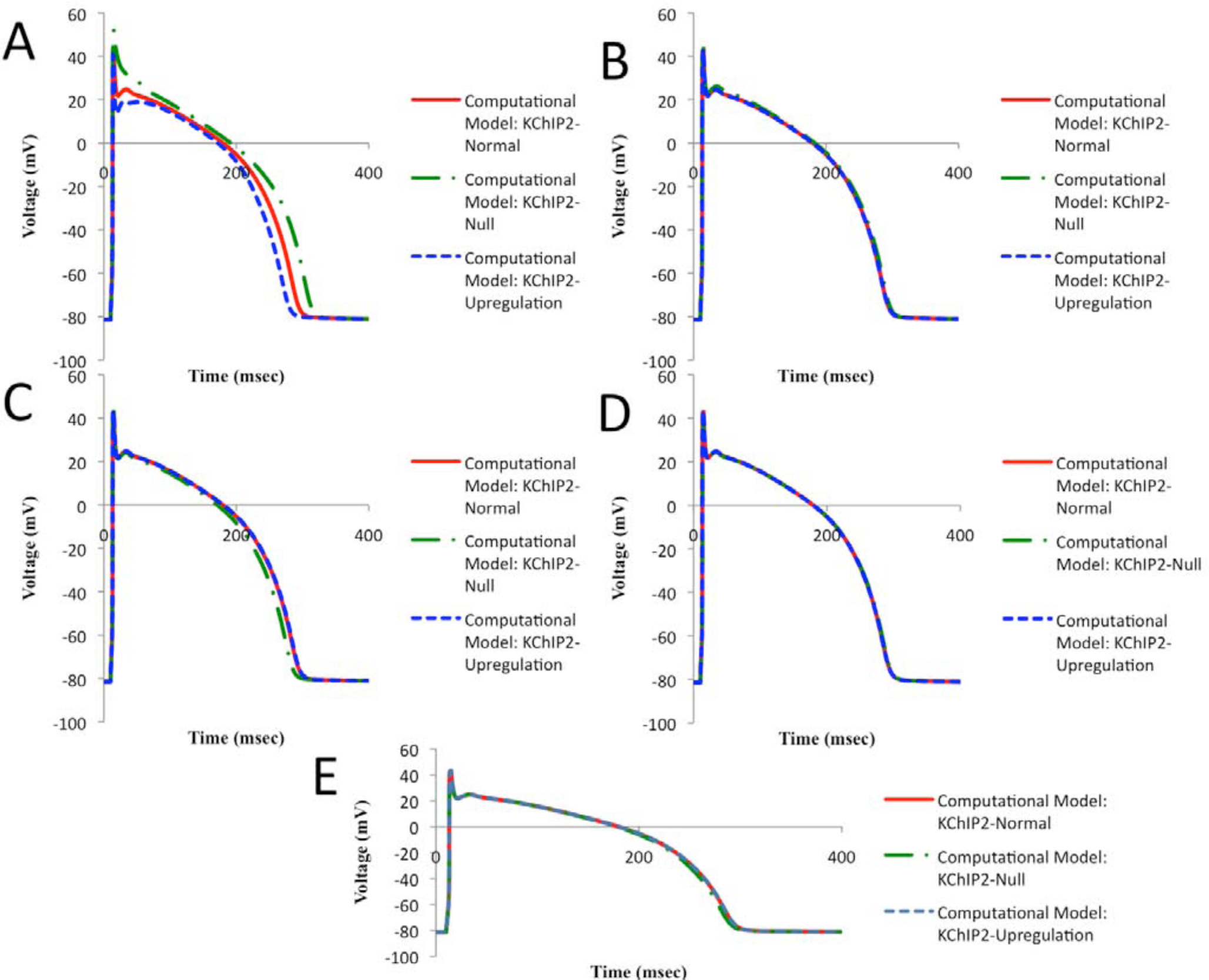
Model predictions on how each current regulated by KChIP2 contributes to the shape of the human ventricular AP. (A) Ito,fast: Resulting APs when the KChIP2 regulating only Ito,fast is set to relatively normal, downregulated, and upregulated concentrations. (B) Ito,slow: Resulting APs when the KChIP2 regulating only Ito,slow is set to relatively normal, downregulated, and upregulated concentrations. (C) ICaL: Resulting APs when the KChIP2 regulating only ICaL is set to relatively normal, downregulated, and upregulated concentrations. (D) INa: Resulting APs when the KChIP2 regulating only INa is set to relatively normal, downregulated, and upregulated concentrations. (E) IKs: Resulting APs when the KChIP2 regulating only IKs is set to relatively normal, downregulated, and upregulated concentrations
Discussion
Our murine computational model accurately reproduces the experimental results recently reported showing how affecting the dynamics of KChIP2 alters the morphology of murine ventricular APs as well as QT interval durations (Jeyaraj et al., 2012). The latter is justified since the duration of the QT interval on the ECG represents the durations of depolarization and repolarization of the ventricles (for further details, see the section on measurement of AP and QT interval durations in the supplementary online material) (Fermini and Fossa, 2003). Specifically, downregulation of the expression of KChIP2 leads to prolongation of AP, QRS, and QT interval durations, and upregulation of the expression of KChIP2 leads to shortage of AP, QRS, and QT interval durations. Furthermore, the duration of the QT interval corresponding to circadian variations over a 24-h period reaches a minimum at around CT12 and a maximum at around CT0. These computational findings agree with experimental results showing that the QT interval is indeed controlled by a circadian clock and presents maximum duration during the first hours of the day (Guo and Stein, 2003; Portaluppi and Hermida 2007; Jeyaraj et al., 2012). Also, our predicted QT interval durations for both models, under relatively normal concentrations of KChIP2, are in accordance with experimentally reported values reported in the introductory section. As seen in Figure 2C, our predicted murine QT interval durations approximate accurately those reported by Jeyaraj et al. (2012). In addition, the magnitude of variation of the human QT interval occurring from our human computational model is between 9 and 23 msec. Bexton et al. (1986) reported a variation of 10-20 msec based on experimental data. Therefore, our model predicts accurately the magnitude of the variation of the human QT interval duration over a 24-h period as well. Moreover, our model predicts that the QT interval durations when KChIP2 is downregulated and upregulated are approximately 392 and 369 msec, respectively. Several studies have reported that QT interval durations higher and lower than those values, respectively, can potentially increase susceptibility to the development of ventricular arrhythmias (Bjerregaard et al., 2010; Kulan et al., 1998).
Deviation from relatively normal concentrations of KChIP2 may lead to the development of cardiac arrhythmias, especially reentrant arrhythmia. The susceptibility to arrhythmias is also increased by aberrant ST segments (Kuo et al., 2001; Thomsen et al., 2009a) and prolonged or shortened QT intervals (and hence APDs). KChIP2 contributes to the regulation of several cardiac ion channels, and therefore deviations from its relatively normal expression can readily lead to a number of channelopathies and hence arrhythmias. In addition, experimental findings (Morganroth et al., 1993; Botstein, 1993; Kulan et al., 1998; Guo and Stein, 2003; Bjerregaard et al., 2010; Jeyaraj et al., 2012) imply that the range of differences in the duration of the QT interval and the ST segment in the normal versus the pathological cases, found with our models, might increase susceptibility to ventricular arrhythmias. Besides the differences in the AP and QT interval durations, KChIP2 affects the upstroke velocities, resting membrane potentials, QRS intervals, and AP-amplitudes predicted by both the computational model and the experimental data provided by Jeyaraj et al. (2012), in the three cases examined, as well. Those differences imply that besides Ito and IKs, other potassium currents are affected by KChIP2 dynamics, such as the inward rectifier potassium current (IK1), which is known to regulate the resting membrane potential of ventricular myocytes (Chilton et al., 2005). Moreover, since INa plays a pivotal role in the rapid depolarization phase of the ventricular AP, and since its density is attenuated during KChIP2 downregulation (Fig. 3C) (Deschênes et al., 2008), the QRS and QT intervals and conduction velocity are expected to be affected as well, thus increasing the susceptibility toward the development of reentrant arrhythmias.
Our murine computational model reproduces with great precision recent experimental results concerning the behavior of KChIP2. Our computational results with the human model have not yet been shown experimentally. However, making predictions about human myocyte electrical activity is more difficult than in mouse. Even though Ito is the major repolarizing current in mice, in humans there are multiple repolarizing currents, and the role of Ito in the formation of the repolarization phase of the AP is uncertain given limited data (Salama and London, 2007; Carbonell et al., 2010; Virág et al., 2011). Also, in humans, Kv1.5 does not encode IKs as in mice, but KvLQT1 does, and the latter has not yet been associated with KChIP2. Finally, the total change in APD predicted by the human model (~20 msec) is less than that predicted by the murine model (~35 msec). This difference may be pronounced because of the shorter APD in the murine model. Nonetheless, it would be interesting to examine whether the KChIP2 dynamics presented here have the same effects on other mammalian models as well.
Computational modeling plays an important role in circadian research and is an important part of research in cardiac electrical activity. We present the first mathematical models of the circadian variation in cardiac electrical activity. These models will be useful partners with experimental validation in future studies. Future work could simulate patches of myocytes rather than individual cells and make more specific circadian predictions about arrhythmia. It also can allow predictions about human cardiac electrical activity, which, of course, is quite important and for which very limited data are available.
Footnotes
Acknowledgements
Conflict of Interest Statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.