
Abstract
Objectives
To characterize the effects of adjunctive brexpiprazole on patient life engagement and depressive symptoms in patients with major depressive disorder (MDD) using patient-reported outcomes.
Methods
An 8-week, Phase 4, open-label, interventional study was conducted at 15 Canadian trial sites between April 2021 and May 2022. Adult outpatients with MDD (at least moderately severe) and inadequate response to 1–2 antidepressants continued their current antidepressant and received oral adjunctive brexpiprazole 0.5–2 mg/day. Co-primary endpoints were change from baseline to Week 8 in Inventory of Depressive Symptomatology Self-Report (IDS-SR) 10-item Life Engagement subscale score, and IDS-SR 30-item total score. Safety was assessed by standard variables.
Results
Of 122 enrolled patients, 120 (98.4%) were treated (mean [SD] dose: 1.2 [0.4] mg/day) and analyzed, and 111 (91.0%) completed the study. Statistically significant least squares mean improvements to Week 8 were observed on IDS-SR10 Life Engagement subscale score (baseline mean [SD]: 16.1 [4.7]; change [95% confidence interval]: −8.11 [−9.34, −6.88]; p < 0.001) and IDS-SR total score (baseline mean [SD]: 41.3 [9.8]; change [95% confidence interval]: −17.38 [−20.08, −14.68]; p < 0.001). Improvements were observed from Week 2, onwards. Treatment-emergent adverse events with incidence ≥5% were fatigue (n = 13, 10.8%), headache (n = 13, 10.8%), insomnia (n = 12, 10.0%), nausea (n = 9, 7.5%), tremor (n = 8, 6.7%), and weight increase (n = 7, 5.8%). Six patients (5.0%) discontinued due to adverse events. Mean (SD) change in body weight from baseline to last visit was +1.9 (3.4) kg.
Conclusions
Using an exploratory patient-reported outcome measure, patients with MDD and inadequate response to antidepressants who received open-label adjunctive brexpiprazole showed early and clinically meaningful improvement in patient life engagement, which should be further assessed in a prospective randomized controlled trial. Patient-rated depressive symptoms (on the validated 30-item IDS-SR) also improved. Adjunctive brexpiprazole was well tolerated, and no new safety signals were observed.
Clinical Trial Registration
ClinicalTrials.gov identifier: NCT04830215.
Introduction
Major depressive disorder (MDD) is characterized by changes in affect, cognition, and neurovegetative functions that significantly impact a person's social and occupational functioning. 1 In addition to improvement of depressive symptoms, patients with MDD value treatment outcomes such as regaining optimism and self-confidence, feeling like their usual selves, and returning to their normal level of functioning. 2 The term “patient life engagement” in the context of psychiatry describes positive life aspects relating to cognition, vitality, motivation/reward, and the ability to feel pleasure, reflecting the functional outcomes of life fulfillment, well-being, and participation in valued and meaningful activities.3,4 Patients with MDD consider life engagement to be relevant and important to their condition. 5
The inclusion of patient-reported outcomes in clinical trials is encouraged by regulatory authorities in order to provide the patient perspective on the effects of treatment. 6 Various patient-reported outcome measures are available to assess aspects of patient life engagement, albeit with differences in scope, psychometrics, and target audience. 7 To address the need for a comprehensive yet specific patient-reported measure of patient life engagement for use in psychiatric clinical trials, expert psychiatrists recently identified 10 items from the Inventory of Depressive Symptomatology Self-Report (IDS-SR) that represent patient life engagement beyond the core symptoms of depression. 8 These items were selected to align with a conceptual framework for patient life engagement, comprising emotional (affect/mood), physical (energy), social (interest) and cognitive (alertness/thinking) domains. 3 The resulting IDS-SR10 Life Engagement subscale has undergone limited psychometric testing, demonstrating content validity via patient interviews, and high internal consistency.5,8
Brexpiprazole is an atypical antipsychotic with serotonergic, dopaminergic and noradrenergic activity 9 that has demonstrated efficacy, safety and tolerability as adjunctive therapy to antidepressant treatment (ADT) in patients with MDD and inadequate response to ADTs.10–13 Post hoc analyses of clinical trial data suggest that adjunctive brexpiprazole may improve patient life engagement from the patient perspective, as evaluated by the IDS-SR10 Life Engagement subscale and from clinical trial exit interviews.3,14 A retrospective real-world database analysis showed that improved patient life engagement was sustained for 6 months following brexpiprazole initiation. 15
The primary objective of this study was to characterize the effects of adjunctive brexpiprazole on both patient life engagement and depressive symptoms in patients with MDD, using patient-reported outcomes. Secondary objectives were to evaluate the effect of adjunctive brexpiprazole on other measures of depressive symptoms, anxiety symptoms, and functioning, and to evaluate the safety and tolerability of adjunctive brexpiprazole in patients with MDD. This study is the first to prospectively investigate the effect of brexpiprazole on patient life engagement, and the first to prospectively use the IDS-SR10 Life Engagement subscale.
Methods
Study Design and Patients
This was a Phase 4, multicentre, open-label, interventional study (ClinicalTrials.gov identifier: NCT04830215; the “ENGAGE” study). The study was conducted in accordance with the International Council for Harmonisation Good Clinical Practice guidelines, the World Medical Association Declaration of Helsinki, and local regulatory requirements. Research ethics board approvals are listed in Supplement 1. All patients provided written informed consent prior to the start of the study.
Patients were enrolled by investigators at 15 clinical trial sites in Canada. Patients were recruited from existing clinic populations, supported by advertisements at three sites. The study followed the requirements of the brexpiprazole Canadian Product Monograph. 16 Key inclusion criteria were: outpatient status; age 18–65 years (or 19–65 years, depending on the provincial age of majority); primary diagnosis of MDD based on Diagnostic and Statistical Manual of Mental Disorders, Fifth edition (DSM-5) criteria 17 ; a current DSM-5 single or recurrent non-psychotic major depressive episode with or without symptoms of anxiety; inadequate response to an adequate trial of 1–2 ADTs (including current ADT) during the current episode per investigator judgment; and a nine-item Patient Health Questionnaire depression scale (PHQ-9) total score of ≥15 (indicating at least moderately severe depression) at the screening and baseline visits. 18 Key exclusion criteria were: previous exposure to brexpiprazole; antipsychotic use within 7 days of baseline; a concurrent DSM-5 diagnosis of schizophrenia, schizoaffective disorder, bipolar I/II disorder, post-traumatic stress disorder, dementia, eating disorder, borderline personality disorder, or antisocial personality disorder; thoughts of death or suicide based on an IDS-SR item 18 score of 3 19 ; or serious risk of suicide in the opinion of the investigator. The following concurrent conditions were permitted, if stable and not the primary focus of treatment: attention deficit hyperactivity disorder, obsessive–compulsive disorder, panic disorder, and generalized anxiety disorder.
The study comprised a screening/washout period of up to 2 weeks followed by an 8-week, single-arm, open-label treatment phase (Figure 1(a)). In the open-label treatment phase, patients continued the stable ADT they were taking at screening (ADT dose changes were not permitted) and were administered adjunctive brexpiprazole orally once-daily with or without food. Brexpiprazole was started at 0.5 or 1 mg/day on Day 1, 16 with up-titration at weekly intervals to 1, 1.5 or 2 mg/day by or at the Week 2 visit, and flexible dosing beyond Week 2 based on clinical response and tolerability (0.5, 1, 1.5 or 2 mg/day). Brexpiprazole tablets were supplied directly to patients by their retail pharmacists through a trial card system.
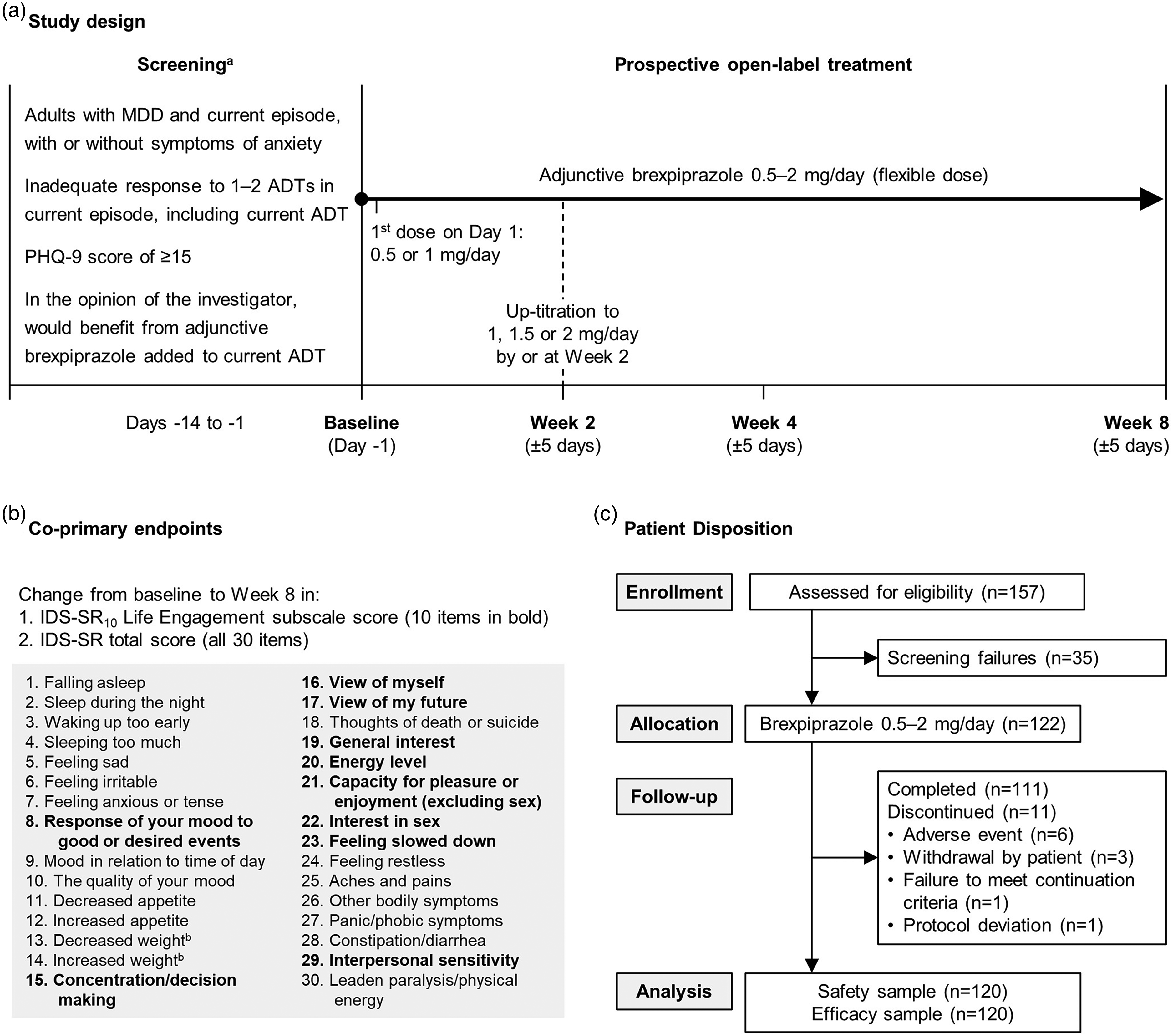
(a) Study design, (b) co-primary endpoints,8,19 and (c) patient disposition. aThe screening visit comprised one or more visits, as needed; screening and baseline visits could occur on the same day if the patient met all required enrollment criteria. bWithin the last 2 weeks. ADT = antidepressant treatment; IDS-SR = Inventory of Depressive Symptomatology Self-Report; MDD = major depressive disorder; PHQ-9 = nine-item Patient Health Questionnaire depression scale.
The study was conducted from April 2021 to May 2022. Patients attended in-clinic visits at Weeks 2, 4, and 8; visits could be remote if needed due to the COVID-19 pandemic.
Benzodiazepines and/or hypnotics (including nonbenzodiazepine sleep aids) could not be ingested in the 8 h prior to a scheduled visit due to the possibility of impacting efficacy assessments. Patients receiving psychotherapy at baseline were permitted to continue psychotherapy provided it remained stable throughout the study.
Assessments
Demographic information, including psychiatric history, was recorded at the screening visit.
Efficacy was assessed using patient-reported measures of depressive symptoms, anxiety symptoms, and functioning, as follows.
The IDS-SR10 Life Engagement subscale comprises a subset of 10 IDS-SR items (items listed in Figure 1(b)) and is scored from 0 to 30, where a higher score indicates lower patient life engagement. 8 Among outpatients with MDD, the minimal clinically important difference is estimated as a 3- to 5-point change from baseline, where 3 points represent a small/moderate improvement and 5 points represent a large improvement. 8
The IDS-SR is a validated measure of 30 core diagnostic depressive symptoms as well as atypical and melancholic symptom features of MDD (items listed in Figure 1(b)).19–21 Each item is scored from 0 to 3, and 28 items are rated (since appetite and weight may increase or decrease, but not both), giving a total score from 0 to 84 where a higher score indicates greater symptom severity. Among in-patients with MDD, an 11- to 15-point decrease in IDS-SR total score corresponds to a clinically relevant improvement (a change in depression severity category from, e.g., severe to moderate, or moderate to mild).20,22
A set of 13 IDS-SR items of specific interest to patients in the context of patient life engagement 5 (described in Supplement 1) was also assessed.
The PHQ-9 comprises one question for each of the DSM-5 MDD diagnostic criteria symptoms and is scored from 0 to 27, where a higher score indicates greater severity of depression.17,18 The Generalized Anxiety Disorder seven-item scale (GAD-7) measures the frequency of bothersome anxiety symptoms and is scored from 0 (best) to 21 (worst). 23 The Sheehan Disability Scale (SDS) measures functional disability across the three items of work/studies, social life, and family life/home responsibilities.24,25 Each item is scored from 0 to 10, where a higher score indicates greater disability; the “Mean” score is the mean of three items (or two items if the patient did not work or go to school). The World Health Organization Disability Assessment Schedule (WHODAS) 2.0 short-form comprises 12 items to assess a patient's activity limitations and participation restrictions; it is scored from 0 to 48, where a higher score indicates greater disability. 26
Two clinician-rated measures were administered to assess overall depressive illness: the Clinical Global Impression–Severity of Illness (CGI-S) and Improvement (CGI-I). 27 The CGI-S is scored from 1 to 7, where a higher score indicates greater severity of illness, and the CGI-I is scored from 1 to 7, where scores of 1–3 indicate improvement, 4 indicates no change, and 5–7 indicate worsening relative to baseline. By definition, a 1-point improvement in CGI-S score or a CGI-I score of 1, 2 or 3 corresponds to a clinically meaningful improvement. 27
Safety was assessed using standard variables, including treatment-emergent adverse events (TEAEs), body weight, and vital signs. Adverse events were assessed by the investigator using the nonleading question, “How have you felt since your last visit?” and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 25.0. TEAEs were assessed by potential causal relationship to trial drug, and by severity (mild, moderate, severe). Serious adverse events (defined in Supplement 1) were also recorded.
Patient diaries were completed at home to assess the clinical experience; diary data are beyond the scope of this article and will be reported separately.
Statistical Analysis
The two co-primary endpoints were change from baseline to Week 8 in (1) IDS-SR10 Life Engagement subscale score and (2) IDS-SR total score. The rationale for using co-primary endpoints was the exploratory nature of the IDS-SR10 Life Engagement subscale (albeit with content validity from patient interviews and high internal consistency 8 ), compared with the more extensively validated 30-item IDS-SR.19–21 The efficacy sample comprised patients who received at least one dose of open-label brexpiprazole and had a baseline and at least one post-baseline efficacy evaluation for IDS-SR10 Life Engagement subscale score and IDS-SR total score. A mixed model for repeated measures (MMRM) with an unstructured variance–covariance matrix was fitted to observed case data with no imputation of missing data. The model included fixed effect terms for visit, baseline value, an interaction term of baseline value by visit, and trial centre. To control the overall type I error at the 0.05 level (two-sided), both co-primary endpoints were required to be significant at the 0.05 level for the study to be considered positive. The sample size calculation is presented in Supplement 1.
Other efficacy endpoints included change from baseline in PHQ-9 total score, GAD-7 total score, SDS Mean and item scores, WHODAS 2.0 short-form score, and CGI-S score, using equivalent MMRM analyses to the primary analysis. CGI-I score was evaluated using a one-sample t-test and last observation carried forward (LOCF) to impute missing data. These endpoints were evaluated at a nominal 0.05 level (two-sided) without adjusting for multiplicity.
Exploratory and post hoc endpoints (anxiety subgroups, correlations, and response/remission) are described in Supplement 1.
The safety sample comprised all enrolled patients who received at least one dose of open-label brexpiprazole. Safety endpoints were summarized using descriptive statistics.
All analyses were performed using SAS Enterprise Guide version 8.1 (SAS Institute Inc., Cary, NC).
Results
Patients
Of 122 enrolled patients, 111 (91.0%) completed the study and 11 (9.0%) discontinued (Figure 1(c)). The most common reason for discontinuation was adverse events; no single adverse event resulted in discontinuation of more than one patient. The efficacy and safety samples comprised 120 patients; two enrolled patients were not assigned brexpiprazole and were excluded from the efficacy and safety samples due to (1) previous exposure to brexpiprazole and (2) suicidality.
Baseline demographic and clinical characteristics are summarized in Table 1. On average, patients had moderately severe-to-severe depression,18,28 moderate anxiety, 23 and markedly impaired functioning 25 at baseline. Of the enrolled sample, 64 patients (52.5%) had a history of psychiatric disorders in addition to MDD, most commonly (≥5%) generalized anxiety disorder (n = 42, 34.4%) and attention deficit hyperactivity disorder (n = 15, 12.3%).
Baseline Demographic and Clinical Characteristics.

Note. Data are mean (SD) except where otherwise stated. a n = 116. b n = 92. c n = 119.
ADT = antidepressant treatment; BMI = body mass index; CGI-S = Clinical Global Impression–Severity of illness; GAD-7 = Generalized Anxiety Disorder seven-item scale; IDS-SR = Inventory of Depressive Symptomatology Self-Report; MDD = major depressive disorder; PHQ-9 = nine-item Patient Health Questionnaire depression scale; SD = standard deviation; SDS = Sheehan Disability Scale; WHODAS = World Health Organization Disability Assessment Schedule.
Ninety patients (75.0% of treated sample) had anxiety symptoms at baseline (GAD-7 total score of ≥10). Patients with anxiety symptoms at baseline had higher mean (standard deviation [SD]) IDS-SR10 Life Engagement subscale score (17.2 [4.3] vs. 13.0 [4.5]) and IDS-SR total score (43.9 [8.6] vs. 33.5 [9.1]) at baseline than patients without anxiety symptoms, indicating lower patient life engagement and greater depression severity, respectively.
All patients continued their ADT from screening, most commonly (>10%) escitalopram (n = 24, 20.0%), venlafaxine (n = 21, 17.5%), bupropion (n = 17, 14.2%), vortioxetine (n = 17, 14.2%), sertraline (n = 15, 12.5%), and citalopram (n = 13, 10.8%).
The mean (SD) brexpiprazole exposure was 56.3 (9.8) days. Most patients started on the lowest 0.5 mg dose: 118/120 treated patients (98.3%) received a 0.5 mg dose at least once in the first 2 weeks of the study. Sixty-two patients (51.7%) received the highest 2 mg dose at some point during the study. The mean (SD) brexpiprazole dose was 1.2 (0.4) mg/day over the entire study (n = 120), and 1.5 (0.5) mg/day during the last 2 weeks of the study (n = 114).
Overall, 535/604 visits (88.6%) were performed in-person, and 69/604 visits (11.4%) were performed remotely.
Efficacy
Statistically significant improvements were observed on the co-primary endpoints of least squares mean change from baseline to Week 8 in IDS-SR10 Life Engagement subscale score (−8.11; standard error: 0.62; 95% confidence interval: −9.34, −6.88; p < 0.001) and IDS-SR total score (−17.38; standard error: 1.36; 95% confidence interval: −20.08, −14.68; p < 0.001) (Figure 2, Table 2). Improvements were observed from the first assessment (Week 2) onwards. Percent improvements from baseline to Weeks 2, 4 and 8, respectively, were larger on the IDS-SR10 Life Engagement subscale (29.6%, 40.1%, and 46.6%) than on the remaining 20 IDS-SR items (20.0%, 26.4%, and 34.6%). Improvement in IDS-SR10 Life Engagement subscale score and IDS-SR total score was observed in both the subgroups with and without anxiety symptoms at baseline and was larger in the subgroup with anxiety symptoms at baseline (Table 2).

Change from baseline in (a) IDS-SR10 Life Engagement subscale score and (b) IDS-SR total score (co-primary endpoints). aConservative estimate (see Assessments section for details). *p < 0.001 versus baseline; MMRM; efficacy sample. ADT = antidepressant treatment; CI = confidence interval; IDS-SR = Inventory of Depressive Symptomatology Self-Report; LS = least squares; MMRM = mixed model for repeated measures.
Summary of Co-Primary and Other Efficacy Results (Efficacy Sample).
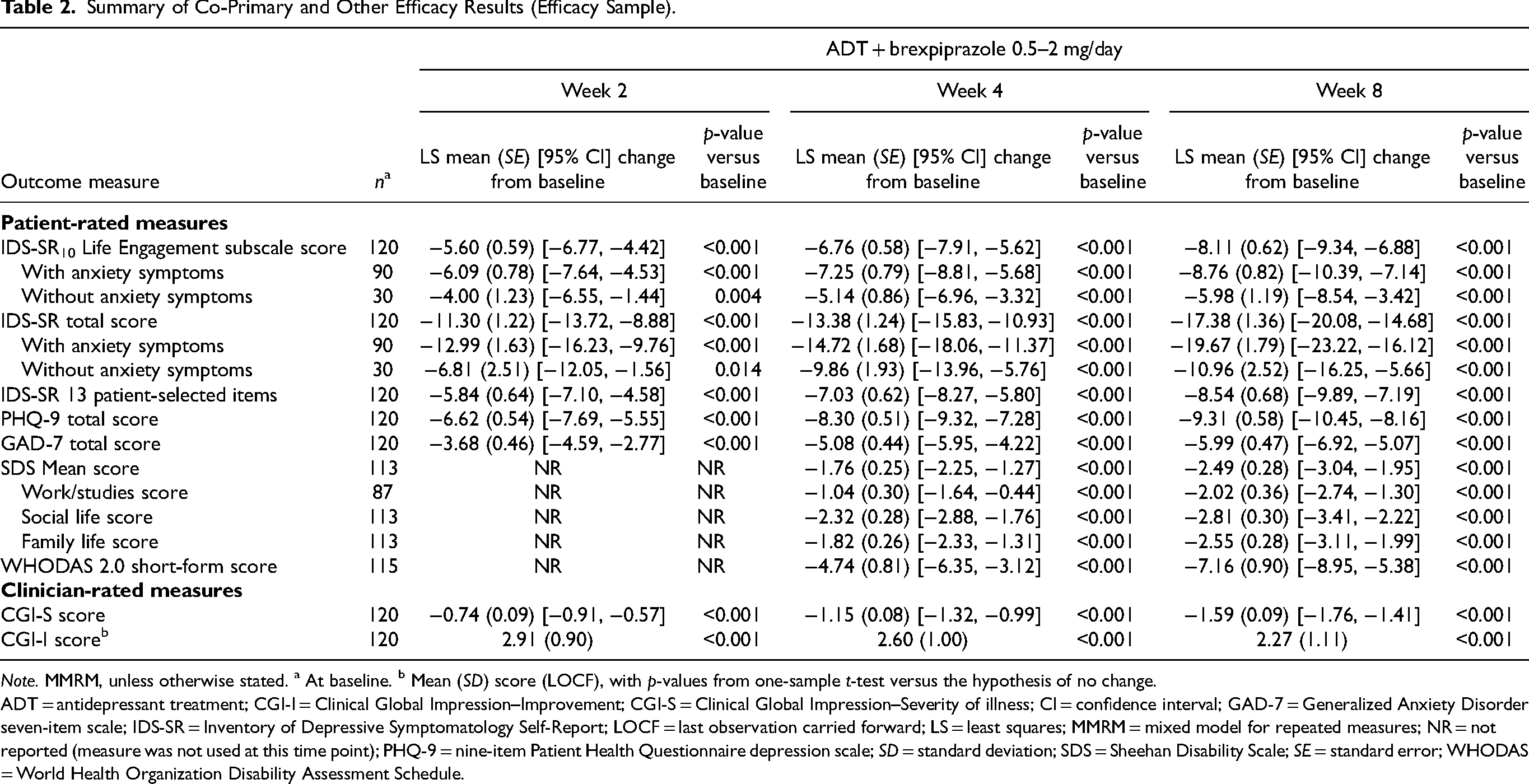
Note. MMRM, unless otherwise stated. a At baseline. b Mean (SD) score (LOCF), with p-values from one-sample t-test versus the hypothesis of no change.
ADT = antidepressant treatment; CGI-I = Clinical Global Impression–Improvement; CGI-S = Clinical Global Impression–Severity of illness; CI = confidence interval; GAD-7 = Generalized Anxiety Disorder seven-item scale; IDS-SR = Inventory of Depressive Symptomatology Self-Report; LOCF = last observation carried forward; LS = least squares; MMRM = mixed model for repeated measures; NR = not reported (measure was not used at this time point); PHQ-9 = nine-item Patient Health Questionnaire depression scale; SD = standard deviation; SDS = Sheehan Disability Scale; SE = standard error; WHODAS = World Health Organization Disability Assessment Schedule.
Mean improvement from baseline to Week 8 was observed on all other efficacy endpoints, including patient-rated measures of depressive symptoms, anxiety symptoms and functioning, and clinician-rated measures of global clinical status (Table 2). Improvements were observed from the first assessment (Week 2 or Week 4, depending on the measurement) onwards.
Significant correlations were observed between patient life engagement and depression, functioning, and disability (presented in Supplement 1). Rates of response and remission increased with time (presented in Supplement 1).
Safety
A total of 211 TEAEs were reported in 81 patients (67.5%). TEAEs with an incidence ≥5% were fatigue, headache, insomnia, nausea, tremor, and weight increase (Table 3). There were no serious adverse events and no deaths. Sixty-three patients (52.5%) had a TEAE considered by the investigator to be related to brexpiprazole treatment. The majority of TEAEs were mild (n = 67, 55.8%) or moderate (n = 35, 29.2%) in severity; 10 patients (8.3%) had severe TEAEs. Four patients (3.3%) reported COVID-19 as a TEAE.
Summary of Treatment-Emergent Adverse Events (Safety Sample).
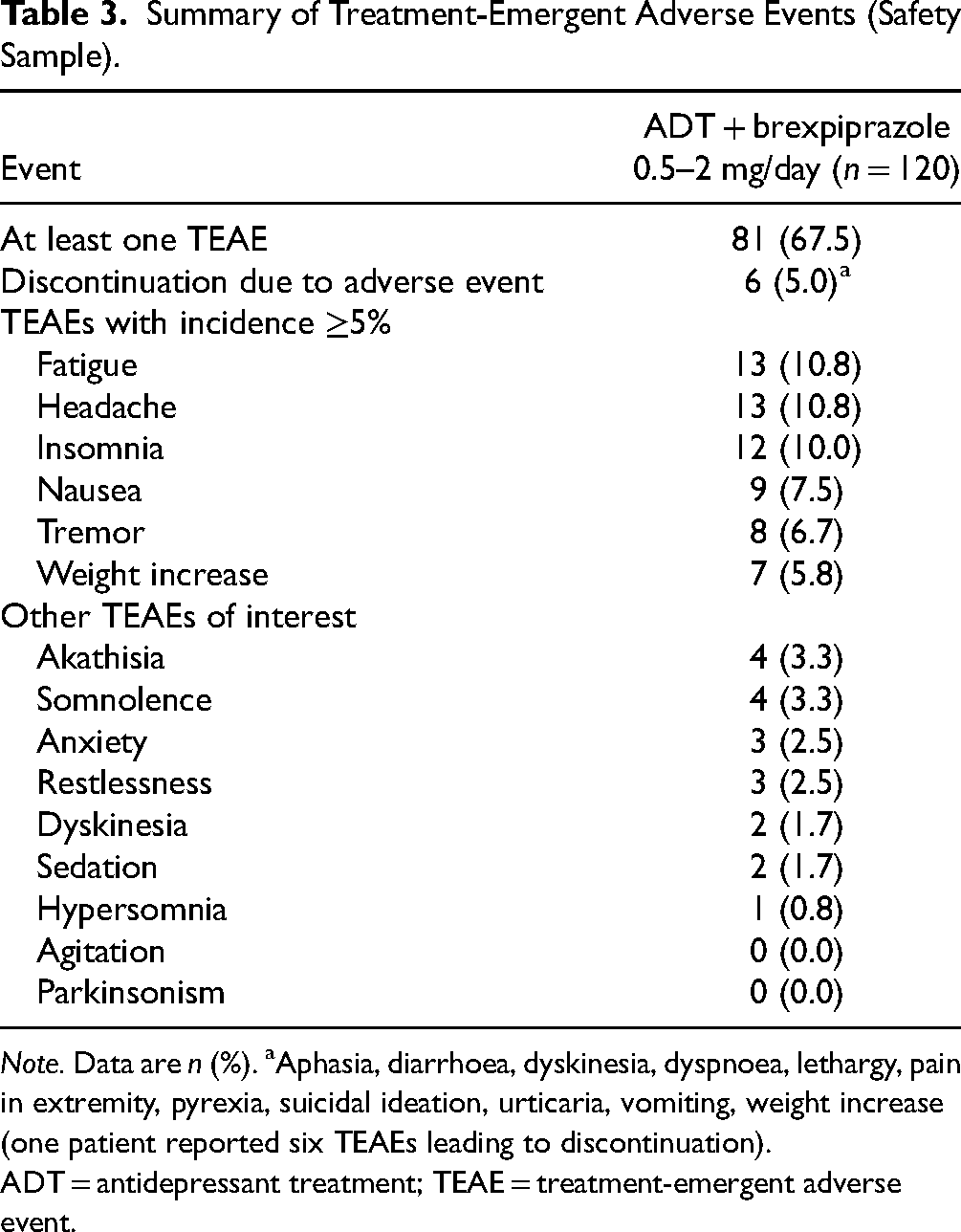
Note. Data are n (%). aAphasia, diarrhoea, dyskinesia, dyspnoea, lethargy, pain in extremity, pyrexia, suicidal ideation, urticaria, vomiting, weight increase (one patient reported six TEAEs leading to discontinuation).
ADT = antidepressant treatment; TEAE = treatment-emergent adverse event.
One patient (0.8%) had a TEAE of suicidal ideation, which was considered by the investigator to be severe and related to study drug. The event occurred on Day 17 of brexpiprazole treatment (current dose, 1 mg). Brexpiprazole was discontinued on Day 23 and a new medication was administered. The patient was withdrawn from the study on Day 32; the event was ongoing, and the patient received continuing care.
Mean (SD) change from baseline in body weight was +1.8 (3.1) kg to Week 8 (n = 101), and +1.9 (3.4) kg to last visit (n = 111). At any visit, 6/111 patients (5.4%) had ≥7% increase in body weight from baseline, and 1/111 patients (0.9%) had ≥7% decrease in body weight from baseline.
Mean systolic blood pressure, diastolic blood pressure, and heart rate remained constant throughout the study (data not shown). At any visit, potentially clinically relevant high values were observed for 3/110 patients (2.7%) for systolic blood pressure, 4/110 patients (3.6%) for diastolic blood pressure, and 4/110 patients (3.6%) for heart rate. No patients experienced potentially clinically relevant low systolic blood pressure, diastolic blood pressure, or heart rate.
Discussion
In adults with MDD and inadequate response to ADT, the administration of adjunctive brexpiprazole was associated with a clinically meaningful, statistically significant improvement from baseline in patient life engagement (IDS-SR10 Life Engagement subscale score), as well as in depressive symptoms (IDS-SR total score, which changed from severe to mild over 8 weeks 28 ). Improvement on both measurements was observed at the first post-baseline visit (Week 2) and continued throughout the study. The improvement in IDS-SR10 Life Engagement subscale score was larger in this study (8.1 points over 8 weeks) than in a previous post hoc analysis of randomized controlled trials of adjunctive brexpiprazole (3.8 points over 6 weeks), 14 which may reflect the open-label nature, real-world patient population and longer duration of the present study. IDS-SR10 Life Engagement scores very strongly correlated with IDS-SR total scores (r > 0.9), as expected for these overlapping measures. However, improvements were larger on the IDS-SR10 Life Engagement subscale than on the remaining 20 IDS-SR items, suggesting that adjunctive brexpiprazole may provide a specific benefit on patient life engagement beyond general antidepressant effects. Overall, this study – the first to prospectively investigate brexpiprazole in patient life engagement and to use the IDS-SR10 Life Engagement subscale – demonstrated an early and meaningful improvement in patient life engagement, as well as in depressive symptoms, among patients with MDD who received adjunctive brexpiprazole.
Patients receiving adjunctive brexpiprazole showed significant improvements on all other patient-rated measures of depressive symptoms, anxiety symptoms, and functioning, at all visits. Depressive symptoms (mean PHQ-9 total score) changed from moderately severe at baseline to moderate by Week 2, and to mild by Week 8, 18 with progressive increases in the proportion with response and remission. The PHQ-9 showed moderate-to-strong correlations with patient life engagement (r = 0.68–0.76), further indicating that, while depression and life engagement are related concepts, patient life engagement is independent from depression to a limited extent (as previously suggested by path analyses 14 ). Anxiety symptoms (mean GAD-7 total score) improved from moderate at baseline to mild at Week 2, 23 with continued improvement to Week 8. Overall functioning (mean SDS Mean score) improved from markedly impaired at baseline to moderately impaired at Week 4 (the first post-baseline assessment of functioning), 25 with further improvement to Week 8. All SDS item scores improved, with a greater improvement on the social life and family life items than the work/studies item, consistent with previous studies showing that improvement of work and school functioning usually lags behind improvement of symptoms.29–31 Patient disability (i.e., activity limitations and participation restrictions as measured by WHODAS 2.0 score) was also reduced. Patient life engagement and functioning/disability are related constructs, and the relationship between improvement of life engagement and improvement of functioning may be bidirectional. 32 Functioning and disability showed moderate-to-strong correlations with patient life engagement (r = 0.50–0.71), providing support for the construct (convergent) validity of the IDS-SR10 Life Engagement subscale.
Clinician-rated measures of overall depressive illness supported the patient-rated outcomes, with clinically meaningful improvement on the CGI-S and CGI-I.
The presence of anxiety symptoms in MDD is associated with worse response to treatment, 33 and poorer patient functioning. 34 In the present study, improvement of patient life engagement and depressive symptoms was observed over 8 weeks in patients with and without anxiety symptoms at baseline. Improvement was greater in patients with anxiety symptoms at baseline than those without anxiety symptoms, as previously observed in trials of adjunctive brexpiprazole, 35 which may reflect the higher mean baseline scores among patients with anxiety symptoms, allowing more room for improvement.
Adjunctive brexpiprazole (mean dose: 1.2 mg) was well tolerated in this study, with a high completion rate (91%), and 5% of patients discontinuing due to adverse events. No new safety signals were observed compared with previous clinical trials in MDD. 36
This study was designed to reflect real-world clinical practice (specifically, clinical practice in Canada) in terms of its open-label nature, patient population, timing of the visits, clinical assessments used, and dosing as per the brexpiprazole Canadian Product Monograph. 16 Certain aspects of study design, including the co-primary outcome measures, were informed by prior semi-structured patient interviews. 5 The majority of the assessments were patient reported; such measures are valuable to gain patient perspectives on treatment effects in domains that are meaningful to patients, and thereby to better inform clinical decision-making and health-care policy. 6
This study is limited by its open-label nature and the lack of a comparator arm. The generalizability of findings may be limited by the exclusion of patients with certain psychiatric comorbidities and suicidality (although enrolment had fewer restrictions than the Phase 3 randomized studies of adjunctive brexpiprazole10–13), and because all trial sites were in a single country (Canada). This was a hypothesis-generating study, and endpoints were tested with no adjustment for multiplicity. The IDS-SR10 Life Engagement subscale was developed based on psychiatrist and patient input in consideration of the four-domain conceptual framework of life engagement; however, the IDS-SR was not originally developed to measure patient life engagement and the subscale may not capture the full scope of patient life engagement. 8 Although the subscale has demonstrated internal consistency, content validity (patient interviews), 8 and now construct validity (correlations), further validation may be warranted, particularly in different socioeconomic and cultural groups and at different stages of depression.
Conclusion
Based on an exploratory patient-reported outcome measure, patients with MDD and inadequate response to ADT who were administered open-label adjunctive brexpiprazole 0.5–2 mg/day showed early and clinically meaningful improvement in patient life engagement – an outcome that is considered relevant and important by patients with MDD, and which should be further assessed in a prospective randomized controlled trial. Patient-rated depressive symptoms (including on the validated 30-item IDS-SR, a co-primary endpoint), anxiety symptoms, and functioning also showed early improvement following adjunctive brexpiprazole treatment. Adjunctive brexpiprazole was well tolerated, and no new safety signals were observed.
Supplemental Material
sj-docx-1-cpa-10.1177_07067437241233965 - Supplemental material for Adjunctive Brexpiprazole for Patient Life Engagement in Major Depressive Disorder: A Canadian, Phase 4, Open-Label, Interventional Study: Brexpiprazole d'appoint pour l'engagement dans la vie des patients souffrant de trouble dépressif majeur: une étude interventionnelle canadienne ouverte de phase 4
Supplemental material, sj-docx-1-cpa-10.1177_07067437241233965 for Adjunctive Brexpiprazole for Patient Life Engagement in Major Depressive Disorder: A Canadian, Phase 4, Open-Label, Interventional Study: Brexpiprazole d'appoint pour l'engagement dans la vie des patients souffrant de trouble dépressif majeur: une étude interventionnelle canadienne ouverte de phase 4 by François Therrien, Caroline Ward, Pratap Chokka, Jeffrey Habert, Zahinoor Ismail, Roger S. McIntyre and Erin M. MacKenzie in The Canadian Journal of Psychiatry
Footnotes
Acknowledgements
The writing support was provided by Chris Watling, PhD, assisted by his colleagues at Cambridge – a Prime Global Agency (Knutsford, UK), and funded by Otsuka Pharmaceutical Development & Commercialization Inc. and H. Lundbeck A/S.
Data Access
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: FT is a full-time employee of Otsuka Canada Pharmaceutical Inc.; CW is a full-time employee of Otsuka Pharmaceutical Development & Commercialization Inc.; PC has served as speaker/received consultant fees from AbbVie, Lundbeck, Janssen, Otsuka, Purdue, and Takeda, and has received research/grant support from AbbVie, Lundbeck, Otsuka, Takeda, Janssen, Telus, and Mitacs; JH has served as a speaker/ad board participant or performed scientific planning committee work for Pfizer, Amgen, AbbVie, GSK, Bayer, Valeo, Boehringer, Eli-Lilly, Elvium, Takeda, Bausch, AstraZeneca, Novartis, Lundbeck, Novo Nordisk, Janssen, Eisai, HLS, Otsuka, Idorsia, MDBriefcase, Liv, MedPlan, Master Clinician Alliance, Academy, Bridge, PeerVoice, Seacourses, Thrombosis Canada, Meducom, CHRC, CTC, STA, CCRN, CPD Network, Telus Health, EOCI, AgenceUnik, Humber Hospital, ABPHE, and CSEM; ZI has received grant support from CIHR, NIH, Brain Canada, and Weston Foundation, and has served as a consultant for Eisai, Lundbeck, Novo Nordisk, Otsuka, and Roche. Additionally, his institution has received funds on his behalf from Biogen and Roche; RSM has received research grant support from CIHR/GACD/National Natural Science Foundation of China (NSFC) and the Milken Institute; speaker/consultation fees from Lundbeck, Janssen, Alkermes, Neumora Therapeutics, Boehringer Ingelheim, Sage, Biogen, Mitsubishi Tanabe, Purdue, Pfizer, Otsuka, Takeda, Neurocrine, Neurawell, Sunovion, Bausch Health, Axsome, Novo Nordisk, Kris, Sanofi, Eisai, Intra-Cellular, NewBridge Pharmaceuticals, Viatris, Abbvie, and Atai Life Sciences. He is a CEO of Braxia Scientific Corp; EMM was a full-time employee of Lundbeck Canada Inc. at the time of this work.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by Otsuka Canada Pharmaceutical Inc. (Saint-Laurent, QC, Canada) and Lundbeck Canada Inc. (Saint-Laurent, QC, Canada), with additional support from Otsuka Pharmaceutical Development & Commercialization Inc. (Princeton, NJ, USA) and H. Lundbeck A/S (Valby, Denmark). The sponsors were involved in the design of the study, the analysis and interpretation of data, the writing and reviewing of this article, and the decision to submit the article for publication.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.