
Abstract
Schizophrenia is a heterogeneous psychiatric disorder that is poorly treated with current therapies. In this brief review, we provide an update regarding the use of animal models to study schizophrenia in an attempt to understand its aetiology and develop novel therapeutic strategies. Tremendous progress has been made developing and validating rodent models that replicate the aetiologies, brain pathologies, and behavioural abnormalities associated with schizophrenia in humans. Here, models are grouped into 3 categories—developmental, drug induced, and genetic—to reflect the heterogeneous risk factors associated with schizophrenia. Each of these models is associated with varied but overlapping pathophysiology, endophenotypes, behavioural abnormalities, and cognitive impairments. Studying schizophrenia using multiple models will permit an understanding of the core features of the disease, thereby facilitating preclinical research aimed at the development and validation of better pharmacotherapies to alter the progression of schizophrenia or alleviate its debilitating symptoms.
Schizophrenia is a severe mental illness with a lifetime prevalence of approximately 0.4% worldwide. 1 It can have devastating effects on patients and their caretakers and imposes enormous costs on health care systems. Schizophrenia is a heterogeneous disorder with positive symptoms (delusions, hallucinations, thought disorders), negative symptoms (anhedonia, avolition, social withdrawal, poverty of thought), and cognitive dysfunction. Although numerous antipsychotic drugs are available to mitigate the symptoms of schizophrenia, the response rate to these drugs is lower than desired, they are slow-acting, and they often produce serious adverse side effects. While the aetiology of schizophrenia is still poorly understood and the biochemical focus has been on dopamine and glutamate, multiple neurotransmitters and neuromodulators, including 5-hydroxytryptamine (5-HT), gamma-aminobutyric acid (GABA), glycine, D-serine, and neuroactive steroids, have been implicated in its pathophysiology. 2 –4 Based on the limited understanding of the biological origins of schizophrenia, there is a continued need for improved animal models of the disorder to better identify the origins of the varied symptoms and to develop and validate novel therapies. This article provides a brief overview of the models currently available and the complexities involved in attempting to develop and use such models.
In general, animal models of complex psychiatric disorders are valuable for studying the neurobiological bases of these disorders and for identifying new drug targets to provide more effective treatment in the future. Preclinical models allow for more rapid monitoring of disease progression than is possible in humans, permit invasive studies of structural and molecular changes across development of the disorder, and allow for relatively easy testing of potential therapeutic agents. 5 However, neuropsychiatric disorders, including schizophrenia, include symptoms such as paranoid delusions and auditory hallucinations that are uniquely human and make interpretation of results obtained from animal models more difficult. 6 For example, increased locomotor activity in response to psychotomimetic compounds such as amphetamine or noncompetitive N-methyl-D-aspartate (NMDA) glutamate receptor antagonists is commonly used as an indication of positive symptoms in schizophrenia, a deficit in prepulse inhibition (PPI, where a weaker sound [prepulse] delivered prior to a stronger sound [pulse] inhibits the natural startle response to the second stimulus) is used as a proxy for sensorimotor gating problems, and changes in social interaction are used as an indicator of negative symptoms. 5 While these tests provide relatively high-throughput evaluation of symptom progression or alleviation with treatment, their relationship to human symptoms can be questioned. More recently, the cognitive symptoms of the disorder have become a focus for research, an important consideration given the lack of effective treatments for these symptoms. The MATRICS initiative of the National Institute of Mental Health (NIMH) identified 7 cognitive domains specifically relevant for schizophrenia, and rodent tests have been proposed for each of these domains. 7 The use of touchscreen-equipped operant conditioning chambers for studying cognition relevant to schizophrenia in rodents is increasingly common given the high face validity, ease of implementation, and high level of automation possible with these tasks. 8 –10 Touchscreen-equipped chambers include a touch-sensitive screen on one wall of the chamber that replaces the levers used in traditional operant conditioning tasks. During the tasks, rodents are required to touch specific areas of the screen according to a given rule to receive the food reward. The NIMH has also proposed the Research Domain Criteria (RDoC) approach for research into the biological underpinnings of behaviour, ultimately including psychopathology and mental disorders. 11,12 RDoC constructs have initially been divided into 5 domains (negative valence systems, positive valence systems, cognitive systems, social processes, and arousal and regulatory systems), all of which are affected in schizophrenia. 13 Work integrating animal research related to mental illness, including schizophrenia, into the RDoC framework is ongoing. 14,15
As discussed in the editorial accompanying this issue, animal models are often sought that satisfy face, predictive, and construct validity. Most of the animal models currently used to study schizophrenia are developmental, drug induced, or genetic. 5 The purpose of this article is to introduce readers of the Canadian Journal of Psychiatry to these strategies for studying schizophrenia in animals with a focus on recent findings. For an in-depth consideration of particular models, comprehensive articles regarding the various animal models of schizophrenia are available. 5,16 –37 Table 1 summarizes common phenotypes associated with animal models of schizophrenia.
Common Phenotypes Associated with Animal Models of Schizophrenia Discussed in the Present Review.
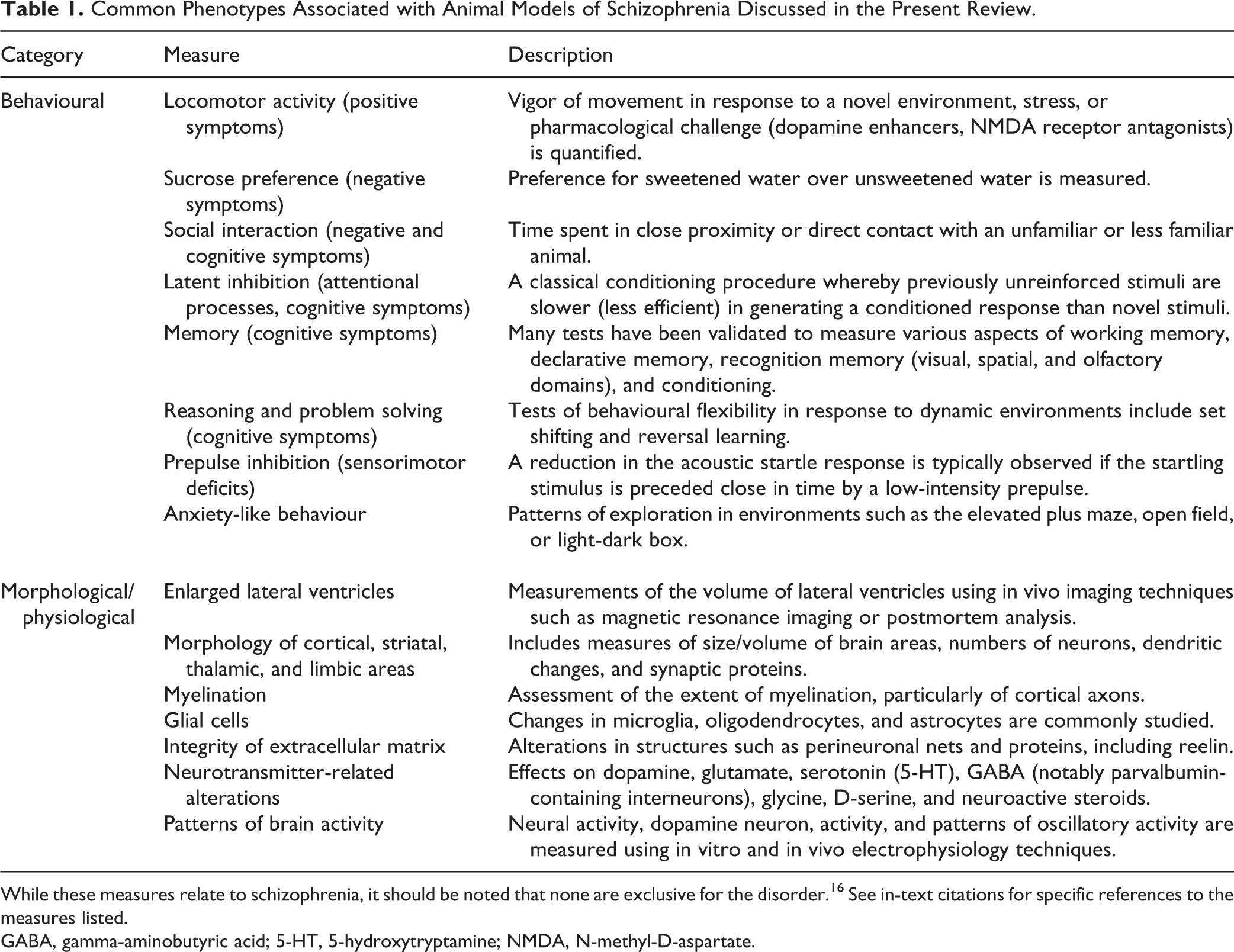
While these measures relate to schizophrenia, it should be noted that none are exclusive for the disorder. 16 See in-text citations for specific references to the measures listed.
GABA, gamma-aminobutyric acid; 5-HT, 5-hydroxytryptamine; NMDA, N-methyl-D-aspartate.
Developmental Models
Although the symptoms of schizophrenia typically appear in adolescence and early adulthood, there is now an extensive body of research linking schizophrenia to prenatal events such as obstetric complications or maternal infection during pregnancy. 38,39 Thus, developmental animal models of schizophrenia involve environmental manipulations and/or drug administration during the perinatal and/or early postnatal period. The offspring are then studied over the course of development. 5 One of the first developmental preparations used to study schizophrenia involved neonatal lesions of the rat ventral hippocampus on postnatal day 7 via local injection of either the excitotoxin ibotenic acid or temporary inactivation via tetrodotoxin. Neonatal lesions, but not similar lesions conducted in early adulthood, lead to an array of abnormalities consistent with schizophrenia that emerge at various times after puberty, 40,41 similar to the typical onset of schizophrenia in late adolescence/early adulthood. Postnatal developmental manipulations involving administration of glutamate receptor agonists/antagonists, postweaning stress, or postweaning social isolation have also been used to model schizophrenia. 33,42 –44 Notably, postnatal social isolation results in behavioural changes in adulthood, including spontaneous locomotor hyperactivity, enhanced response to novelty, deficits in sensorimotor gating, cognitive impairment, and increased anxiety and aggression. 5
The methylazomethanol (MAM) model involves prenatal administration of MAM into pregnant rats. 45,46 MAM is an antimitotic and antiproliferative agent that methylates DNA 47 and acts specifically on proliferation of neuroblasts without affecting glia or producing teratogenic effects in peripheral organs. 48 Administering MAM to pregnant rats results in neuroanatomical, electrophysiological, and behavioural changes in the offspring that depend on the gestational day (GD) of administration, with GD17 animals being the most commonly used in schizophrenia animal models. 49 –51 Changes in MAM offspring include decreased volume of the medial prefrontal cortex, increased volume of lateral and third ventricles, decreases in parvalbumin interneuron markers in limbic and cortical areas, increased dopamine neuron activity in the ventral tegmental area, increased locomotor response to amphetamine and MK-801, decreased social interaction, decreased PPI, impaired memory, and increased anxiety (reviewed in Moore et al. 45 ).
A second prenatal developmental strategy involves inducing maternal immune activation (MIA) in pregnant rodents with agents like the viral mimetic polyinosinic-polycytidylic acid (poly I: C) and studying the consequences for the offspring. 52 –62 A rather large body of evidence indicates that the offspring of rodents and primates that have undergone MIA during pregnancy show anatomical, neurochemical, electrophysiological, and behavioural changes consistent with schizophrenia, 63 –69 as well as other neurodevelopmental disorders, including autism. 70 The maternal immune response appears critical for the effects of MIA on the offspring, with roles for maternal interleukin-6 71 and interleukin-17a 72,73 identified. Recent work using the model has identified novel pathological changes in the offspring. For example, recent findings using this animal model implicate changes in parvalbumin interneurons and their associated perineuronal nets (extracellular matrix structures involved in structural and synaptic plasticity) in the prefrontal cortex (PFC) and hippocampal formation in cognitive deficits, 69,74,75 consistent with studies on postmortem tissue from individuals with schizophrenia. 74 –76 Using mice, Shin Yim and colleagues 77 showed that abnormalities in the primary somatosensory cortex underlie behavioural changes in the offspring following MIA.
Neurodevelopmental models permit investigations of the processes leading to the development of schizophrenia during the prodromal phase of the disease and potential development of prophylactic treatments to prevent the progression to psychosis. 78 Combined, recent work in the MAM, MIA, neonatal ventral hippocampal lesion, and postweaning stress and isolation models has shown that interventions conducted around the time of adolescence alleviate some of the brain and behaviour changes in the animals. 79 –85 These interventions include cognitive training, antipsychotic administration, antioxidant administration, and benzodiazepine administration. Moreover, these models allow unique opportunities to create “2-hit” models, 86 in which development is challenged during 2 critical periods (e.g., early brain development and then adolescence) in an effort to trigger a phenotype consistent with the development of schizophrenia later in life. While a single model is not sufficient to confirm efficacy for novel therapies, these well-validated neurodevelopmental models replicate many of the behavioural and neuroanatomical phenotypes of schizophrenia. 87,88 Each of these components of schizophrenia can then be targeted for pharmacotherapy independently with separate molecules or specifically designed multitarget drugs, 89 with the models providing accurate readouts of treatment efficacy.
Drug-Induced Models
The drugs used most frequently in animal models of schizophrenia are dopamine enhancers (e.g., amphetamine and apomorphine) and noncompetitive NMDA receptor antagonists (phencyclidine [PCP], ketamine, and MK-801). Amphetamine-induced psychosis in humans is well known and is proposed to result from the increased synaptic dopamine produced by this drug. Repeated, intermittent administration of amphetamine in rodents results in a persistent sensitization typified by exaggerated hyperactivity in response to an acute amphetamine challenge. 90 However, repeated amphetamine does not induce social interaction deficits (which are proposed to be equivalent to negative symptoms in schizophrenia) 91 and appears to affect only some cognitive tasks related to PFC function while leaving hippocampal function relatively unaffected. 90 In summary, amphetamine induces psychotic-like changes but does not reliably produce negative or cognitive symptoms. In fact, some have proposed that treating selected patients with psychostimulants in an effort to improve negative symptoms is worth exploring. 92
Hypofunctioning of NMDA glutamate receptors in schizophrenia has received considerable interest in recent years. Hence, noncompetitive NMDA receptor antagonists (ketamine, PCP, MK-801) have been administered to rodents in the hope that they would provide useful animal models of schizophrenia. 93 –98 In humans, PCP induces hallucinations and delusions, progressive withdrawal and poverty of speech, and impaired cognitive performance. 99 –101 In rodents, acute PCP produces hyperlocomotion (proposed to have translational relevance to positive symptoms in humans), social withdrawal, impaired PPI, and cognitive deficits. 102 –104 Effects of new antipsychotic candidates such as positive allosteric modulators of metabotropic glutamate receptors, the tetrahydroprotoberberine govadine, and nitric oxide donors (e.g., sodium nitroprusside [SNP]) have been tested using acute MK-801–induced disruptions of touchscreen-based tasks relevant to schizophrenia. These novel treatments reverse deficits on tasks, including paired associates learning and trial-unique, delayed nonmatching-to-location tasks. 105 –108
NMDA receptor activation results in the production of neuronal nitric oxide (NO). Bujas-Bobanovic and colleagues 109 studied this relationship and found that PCP-induced effects in rats were blocked by the NO donor SNP. Hallak et al. 110 translated these findings to patients and found that SNP, when administered by intravenous (IV) perfusion to patients with schizophrenia undergoing antipsychotic drug therapy, showed signs of improvement on scales measuring symptom severity within 4 hours. Notably, the improvements persisted for at least a week after a single dose of SNP. In healthy subjects, SNP was able to prevent psychogenic symptoms induced by ketamine. 111 Other studies by the same group in rats showed that a single dose of SNP prevented psychosis-like behaviour produced by ketamine 112 and that SNP treatment more consistently decreased ketamine-induced behaviours than other NO-related compounds. 113 Thus, in these SNP studies, basic science research findings in animal models were translated to the clinical situation, then reverse translated back to animal models to further investigate mechanisms of action and other potential therapies. Such bidirectional translation is essential to developing new insights into the pathophysiology of schizophrenia and new avenues for treatment.
There has been a great deal of interest in the possible role of the amino acids glycine and D-serine, both coagonists at the NMDA receptor, in the aetiology and pharmacotherapy of schizophrenia. D-serine has a higher affinity than glycine for the serine glycine binding site on the GluN1 subunit of the NMDA receptor, and the focus of research has been more on D-serine in recent years. This emphasis will no doubt increase with recent withdrawal of glycine reuptake inhibitors from clinical trials. 114 D-serine is formed from L-serine via serine racemase and catabolized by D-amino acid oxidase (DAO), and there is a great deal of interest in studies in rodents in which these enzymes are inhibited or induced by drugs or are knocked out, knocked down, or overexpressed in transgenic mice 115,116 in an effort to identify new drugs that may be effective antipsychotics. Genetic studies on D-serine will be discussed in a later section of this review.
Other models have used local injections of drugs into specific brain areas in an effort to mimic a known pathophysiology associated with schizophrenia. For example, alterations of parvalbumin-containing GABAergic interneuron number and function in the prefrontal cortex is strongly associated with schizophrenia 117 and a number of animal models of the disorder. 74 Generally, these alterations are thought to result in reduced output of these neurons, thereby disturbing circuit function. In an effort to mimic this state in rodents, Tse and colleagues 118 microinjected the GABA-A receptor antagonist bicuculline into rodent medial PFC and assessed cognition. Alterations in attentional processes, working memory, speed of processing, cognitive flexibility, emotion regulation, and locomotor activity consistent with a schizophrenia-like phenotype were observed.
Genetic Models
A genetic contribution to schizophrenia has long been a focus of study and the basis for several animal models. Numerous candidate genes have been associated with an increased risk of developing schizophrenia, and most of these genes relate to proteins relevant to neuronal plasticity, glutamatergic or dopaminergic function, and synaptogenesis. 118 –120 The largest genome-wide association study thus far, including data from 37,000 patients and 113,000 controls, was published in 2014. 121 From this data set, 108 loci associated with schizophrenia were identified. Many of these genes have been used to generate animal models of schizophrenia, and new loci will allow for more models in mice. Notably, the identification of candidate genes is the first step in the generation of more sophisticated animal models of schizophrenia that incorporate the complex gene-by-environment interactions and gene-gene interactions that underlie the disease in humans. 122 Several of the most established models based on genetic mutations are discussed below.
DISC1 (disrupted-in-schizophrenia 1) is a gene for DISC1, a synaptic protein expressed early in development and involved in pre- and postnatal development in neurons. DISC1 is considered a highly penetrant mutation and a risk factor for schizophrenia, although the genetic evidence for this claim has been debated. 123 Mice with inducible and/or partial loss of DISC1 function have brain morphology consistent with schizophrenia, including enlarged ventricles and reduced cortical thickness and brain volume. 124 Reduced immunoreactivity for parvalbumin, a marker of fast-spiking inhibitory interneurons, has also been observed in the medial PFC and hippocampus, consistent with postmortem studies in humans. 124 While hyperactivity and impairments in volitional behaviour have been observed, there is some controversy about the degree to which these animals show changes in PPI, locomotor activity, or differences in negative symptoms and cognitive abilities. 125 –128
22q11.2 deletion syndrome is a genetic syndrome resulting from a deletion on chromosome 22q11.2. 129 These deletions are associated with schizophrenia in humans and have been used to generate animal models of schizophrenia. It has been estimated that 22q11.2 deletions account for approximately 1% of cases of schizophrenia, with some studies suggesting as many as 22.6% of those with the deletions are diagnosed with schizophrenia or schizoaffective disorder. 130 The core symptoms, treatment response, cognitive deficits, and brain pathologies do not appear to differ in 22q11.2 deletions relative to those diagnosed with schizophrenia that do not have the deletions. The mouse model recapitulates the major neuroanatomical findings observed in 22q11.2 deletion carriers, 131 with alterations in cortical and subcortical grey matter volumes, enlarged ventricles, and reduced spine size and dendritic complexity. 132 Behavioural studies in mice with 22q11.2 microdeletions reveal deficits in PPI, fear conditioning, impaired spatial memory, and deficits in delayed nonmatching to place tasks. 133
Mutations in the genes for the cell adhesion molecule neuregulin 1 (NRG1) and its receptor ErbB4 also confer increased risk for schizophrenia. 134 –136 The NRG1 gene regulates expression of several isoforms of NRG1 that are involved in the development of the nervous system. 137 Homozygous knockout (KO) in mice is lethal, but viable heterozygous, hypomorphic conditioned KOs can modulate neuregulin-ErbB4 signaling. 138 These mice exhibit a behavioural phenotype with increased activity and exploration, and some reports noting working memory impairments in tests of spontaneous alternation 138,139 in NRG1 hypomorphic mice, but not Erb4 hypomorphs, show reduced PPI. Notably, NRG1 hypomorphs have significantly fewer functional NMDA receptors than wild-type mice and reduced dendritic spine density on pyramidal neurons of the hippocampus.
Dysbindin is a synaptic protein implicated in the regulation of exocytosis, vesicle biogenesis, and receptor trafficking processes involved in excitatory synaptic transmission. 140 A reduction in dysbindin expression affects NMDA receptor function and impairs working memory. Genetic variations in the gene encoding dysbindin impair cognition in humans and increase the risk for schizophrenia. 140 –145 Moreover, dysbindin gene and protein expression are reduced in the hippocampus and PFC of schizophrenic patients. Dysbindin mutant mice show behaviours consistent with a schizophrenia-like phenotype, including hyperactivity, learning and memory deficits, and increased impulsive and compulsive behaviours. Moreover, the mutants have disrupted dopamine/D2 signaling and altered neuronal excitability. 146 –149
Reelin is involved in synaptic formation and plasticity, and reduced reelin messenger RNA (mRNA) and its corresponding protein have been reported in cerebellum, hippocampus, and frontal cortex of individuals with schizophrenia. 5 Deletion of 1 allele encoding the reelin gene produces a heterozygous mutant mouse. 150 These mice have some schizophrenia-like pathology, including increased neuronal packing and reduced dendritic spine density in the frontal cortex and hippocampus. 151 However, there is considerable controversy about the behavioural relevance of this model to schizophrenia, with variable findings in studies of social interaction, cognitive deficits, locomotor response, and PPI. 5
Several genes showing up in association studies on schizophrenia are related to D-serine and include G72 (or DAP activator), DAO, and SRR (the gene for serum racemase). 115,116 Insertion of the human G72 gene in mice produces a model that mimics some behavioural and cognitive characteristics of schizophrenia. 152 The genes for the enzymes involved in the formation (serine racemase) and catabolism (DAO) of D-serine can be manipulated relatively easily. Behavioural effects noted in mice in which serine racemase is inhibited (leading to decreased brain levels of D-serine) indicate that this situation results in behavioural effects corresponding to symptoms in humans with schizophrenia. 153 Conversely, genetic inactivation of DAO enhances extinction and reversal learning, suggestive of improvement of schizophrenia-like symptoms. 154 It should also be mentioned that research now indicates that D-serine has antidepressant properties and may be a biomarker for nonresponse to the antidepressant effects of ketamine. 155,156 Similarly, there is now extensive literature supporting GABAergic dysfunction in schizophrenia, including its interactions with dopamine and some neuroactive steroids. 157 With regard to animal models, deficiency of glutamate decarboxylase 67, the enzyme responsible for the conversion of glutamate to GABA in mice, results in a schizophrenia-like phenotype. 158
Sex Differences in Animal Models of Schizophrenia
There are clear sex differences with respect to age of onset, symptoms, and treatment/drug response in schizophrenia. 159 Sex differences also exist between males and females in animal models of schizophrenia. 160,161 These differences have been detected in both neurodevelopmental and genetic models of schizophrenia. Male- or female-specific deficits in PPI in offspring after prenatal PolyI: C have been reported, while deficits in both sexes were found in other studies. 162,163 Similarly, male-specific deficits have been reported in studies of fear conditioning, 164 set shifting, 165 and recognition memory, 166 while female-specific impairments have been reported in some, but not all, studies of spatial memory in polyI: C offspring. 167 In general, prefrontal cortex–dependent tasks, including latent inhibition, 168 PPI, 169 recognition memory, 170 and social behaviour, 162 tend to be impaired to a greater extent in males than in females after MIA. These sex differences may parallel altered glutamate, aspartate and taurine concentrations, and parvalbumin-expressing inhibitory interneuron cell density in the prefrontal cortex of males but not females. 168 Interestingly, MIA with polyI: C may preferentially disrupt prefrontal cortex–dependent tasks, and these deficits may only manifest in adulthood, potentially due to the delayed maturation of prefrontal cortex or puberty-related hormonal changes. 160 However, statistical power (small group sizes per sex) and timing of polyI: C injection vary between studies and are important considerations in interpreting variable sex differences between polyI: C studies. 160 Sex differences have also been reported in a neurodevelopmental model using prenatal exposure to the bacterial toxin lipopolysaccharide (LPS). Wischhof et al. 57 reported that males, but not females, exhibited prolonged PPI deficits and impaired recognition memory. Interestingly, this study also reported deficient myelination in corticolimbic areas and reduced parvalbumin expression in the prefrontal cortex and hippocampus.
Overall, data on sex differences in genetic models of schizophrenia in the literature are variable and dependent on adequate sample size. 160 For example, studies investigating mutations in DISC1 have found that when mutations are induced postnatally, sex-specific behavioural differences are observed, with males exhibiting hyperactivity and altered social interaction while females are impaired in spatial memory. 170 Pre- and postnatal mutant expression results in increased aggression, altered social behaviour, and sensitivity to psychostimulant-induced locomotor hyperactivity in males and more depressive phenotypes in females. 171 Male-specific increases in the ratio of excitatory to inhibitory synaptic activity in cortical pyramidal cells have also been reported in DISC1 mutants. 172 Notably, mice with a naturally occurring mutation in DISCI show a similar phenotype to the inducible mutants, with male-specific hyperactivity. However, in these mutants, it was males that exhibited a depressive phenotype. 173 This variability is common in studies of DISC1, and approximately half of studies to date have reported no sex differences in cognitive tasks, depressive behaviours, or locomotor activity. 154
Variable sex differences have also been observed in other genetic models. Overall, cognitive impairments tend to be more severe in males in NRG1-ErbB4 mutants. 160 Male-specific deficits in spatial learning in the Morris water maze have also been reported, 174 but many studies lack statistical power to adequately detect sex differences. 160 As such, further investigation of sex-specific effects on mutations affecting NRG1-ErbB4 is required. While most studies including both sexes of reelin mutants lacked sufficiently large samples to differentiate between sexes, differences have been observed in studies of heterozygous reelin mutant mice with larger samples. 175 Adult male, but not female, heterozygous reelin mutant mice have enhanced locomotor response to MK-801 175 as well as deficits in social recognition memory. In virtually all cases, additional studies with adequate sample size for both sexes are required to better define sex differences in genetic models.
Glial Contributions to Schizophrenia
Glial cell dysfunction has been implicated in several animal models of schizophrenia. 176 –178 Activated microglia are an important part of the immune system and, by releasing proinflammatory cytokines, may contribute to symptoms seen in animal models based on immune reactions. 54 –56,179 Genetic mutation of the genes encoding CX3C chemokine receptor 1 (CX3CR1) fractalkine (CX3CL1) and its receptor (CX3CR1), which are concentrated in microglia and involved in immune cell trafficking to the central nervous system, induces a schizophrenia-like phenotype. 180,181 Similarly, genetic and proteomic studies in humans have suggested that abnormalities in the cytokines interleukin-6, interleukin-1B, interferon-Y, tumour necrosis factor α, and the major histocompatibility complex are associated with schizophrenia. 182 –186 Genetic and transcriptomic studies of S100B and glial fibrillary acid protein, both highly expressed in astrocytes, reveal altered expression in the plasma and brains of individuals with schizophrenia. 187,188 There is now also substantial evidence, using demyelination animal models such as rodents treated with cuprizone, that oligodendrocyte function and demyelination may also contribute to the symptoms of schizophrenia. 189,190 Oligodendrocyte-related genes are frequently identified as associated with schizophrenia in genetic, transcriptomic, and proteomic studies. 191 Consistent with this, a genomic analysis of 10 publicly available gene sets identified a glial-oligodendrocyte pathway as significantly associated with schizophrenia, suggesting that these myelination abnormalities in schizophrenia are at least in part due to inherited factors. 192 Thus, modeling of schizophrenia involves the integration of genetic factors across multiple cell types, neurodevelopmental processes, and complex gene-by-environment interactions.
Summary
Despite the complicated, heterogeneous aetiology of schizophrenia, which almost certainly includes contributions from both environmental and genetic factors, animal models have increased our knowledge of the brain pathology in schizophrenia. Moreover, these models allow for identification of pathways for pharmacotherapy and improved screening and validation of potential novel antipsychotics. With continued refinement of the models and our understanding of the pathophysiology of schizophrenia, the development of more rapidly acting therapies with reduced side effect profiles compared to the agents currently available is possible.
Footnotes
Acknowledgements
The authors are grateful for funding from the University of Alberta, the University of São Paulo, and the University of Saskatchewan. John G. Howland’s research in this area is supported by operating grants from the Canadian Institutes for Health Research (125984 and 153111). Ian R. Winship’s research in this area is supported by operating grants from the Canadian Institutes for Health Research (153111), the Natural Science and Engineering Research Council (RGPIN-2017-05380), the University Hospital Foundation, and Alberta Innovates Health Solutions.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.