
Abstract
Objective:
This study aimed to evaluate whether EDTA-based targeted chelation therapy can act as senomorphic in chronic kidney disease (CKD)-induced vascular calcification.
Introduction:
Vascular calcification, a significant complication of CKD, is induced due to osteogenic trans-differentiation and senescence of vascular smooth muscle cells (VSMCs). Senescent VSMCs contribute to inflammation and calcification via the senescence-associated secretory phenotype (SASP). Recent evidence implicates the NLRP3 inflammasome as a key mediator of inflammation and senescence in vascular calcification. We previously demonstrated that EDTA chelation therapy removes calcium deposits from arteries in the CKD model. In this study, we investigated whether EDTA also exerts senomorphic effects by reducing NLRP3 expression and vascular cell senescence in calcified aortic tissue.
Methods:
We used an adenine diet-based rodent model of late-stage CKD and an ex vivo aortic ring culture model to evaluate the senotherapeutic potential of EDTA-loaded human serum albumin nanoparticles tagged with anti-elastin antibody—Flexibzumab (EDTA NPs). For validation, we performed a comparative proteomics analysis on the total proteins harvested from the abdominal aortas of the EDTA-treated and untreated animals.
Results:
Our results show that targeted chelation therapy with EDTA NPs decreases the percentage of SA-β-gal positive senescent cells in the calcified aorta and acts as senomorphic by decreasing NLRP3 inflammasome formation, which is a primary intracellular source of senescence-associated secretory phenotype (SASP).
Conclusion:
For the first time, the current study provides proof of concept on the senotherapeutic potential of a targeted chelation therapy and its capacity to modulate SASP from the senescent cells accumulated in calcified aorta.
Keywords
Highlights
Our findings show that chelation therapy can act as senomorphic and increase rodents’ life span from heavy vascular calcification.
Chelation therapy decreases senescent cell accumulation, SASP and NLRP3 expression in the aorta.
Chelation therapy is a novel method for reprogramming senescent cells in the aorta to prevent their phenotypic switching to inflammatory senescent cells and ultimately to osteoblasts.
Current data have provided a new hypothesis that agents that restore imbalance in the cellular microenvironment (in this case, EDTA) have the potential to act as senomorphics, which can serve as safer therapeutic alternatives over senolytics to treat vascular calcification by decreasing apoptosis.
Introduction
Aging-associated medial artery calcification (MAC), also known as Mönckeberg’s calcification, is the most common form of calcification that is observed during aging and is a major contributor to cardiovascular morbidity and mortality. The aging process is typically accelerated in patients with chronic kidney disorder (CKD), as is evidenced by progressive vascular disease, persistent uremic inflammation, muscle wasting, osteoporosis, and frailty that precedes terminal renal failure.1,2 Although the exact underlying mechanisms of accelerated vascular aging in CKD have not yet been fully elucidated, many preclinical studies emphasize the critical role played by the increased number of senescent cells that accumulate in the calcification of the aorta. Recently, Fang et al. 3 reviewed the role of cellular senescence during vascular calcification in CKD. Oh et al. 4 have demonstrated the presence of senescent cells (as defined by p16High and p53 positive expression) in the aortic valves of individuals suffering from calcified aortic valve disease, and that the severity of tissue remodeling in calcific aortic valve disease is proportional to the expression of P16INK4A. Preclinical data from the labs of leaders in the field, Judith Campisi and James Kirkland, have supported the view that senolytics—agents that cause selective apoptosis of senescent cells-can be a potential therapy for treating vascular stenosis by clearing senescent cells from atherosclerotic plaques.5–8 These data support the compelling view that senescent cells play a critical role in vascular calcification and hence justify the hypothesis that senolytics (agents that induce selective apoptosis of senescent cells) and senomorphics (agents that alter Senesce associated secretory phenotype from the senescent cells and modulate the local immune environment without inducing their apoptosis) could provide an attractive therapeutic strategy to prevent and treat vascular calcification.
Vascular calcification is accompanied by a severe pro-inflammatory response that exacerbates disease progression.9,10 The highly inflammatory secretory phenotype from senescent (SASP) cardiovascular cells has been reported to induce osteogenic changes in the aortic VSMCs.11,12 NLRP3 inflammasome activation in the cardiac cells has been identified as a key source of cellular senescence and SASP, leading to premature cardiac aging and progression of cardiovascular disorders.13–15
Multiple upstream pathways have been reported to cause activation of NLRP3 inflammasome signaling during vascular calcification. Direct stimulation by Calcium crystals is one of the most direct and vital mechanisms that causes activation of the NLRP3 inflammasome in the calcified aorta. Upon activation, the NLRP3 inflammasome undergoes self-oligomerization and rescues apoptosis-associated speck-like protein (ASC) from degradation. ASC is a death domain family protein with a Caspase 1 Activation and Recruitment domain. ASC rescue turns on the caspase-1 effector pathway, leading to conversion of pro IL-1β to IL-1β, and pro-caspase-1 to caspase-1 and ultimately the release of active caspase-1, IL-1β and IL-18, causing amplification of inflammatory responses as well as pyro-apoptosis.16–18
We have previously demonstrated that chelation therapy with EDTA-loaded nanoparticles specifically targeting degraded elastin reversed existing heavy mineral deposits in arteries. The reversal of aortic calcification was accompanied by a significant reduction of bone-associated mRNA expression of BMP2 and RUNX2, reflecting that chelation therapy and organic removal of calcium deposits have the potential to prevent phenotypic transition of VSMCs to osteoblasts at the genetic level.19–21 BMP2 is a key transcription factor that regulates the expression of both p21 and RUNX2 at the transcriptional level. 22 Prompted by this preclinical evidence, for the current study, we analyzed the impact of EDTA-based chelation therapy on the senescence markers and the NLRP3 expression, identified as a potential source of SASP.23–25
We employed an in vitro aortic ring culture model and an in vivo chronic kidney disease model to investigate the impact of chelation therapy on the accumulation of senescent cells in the aorta. The samples were tested for the presence of senescence and SASP markers and for NLRP3 expression in the aorta. Human serum albumin nanoparticles loaded with EDTA (EL-EDTA NPs) were conjugated with Flexibzumab antibody that targets explicitly damaged elastin in the artery.19,21,26 The EL-EDTA-NPs were injected IV in the late-stage CKD animals, and aortas from the animals were harvested to analyze calcification status, senescence phenotype, and NLRP3 expression. Overall, this study provides proof of concept for using chelation therapy as a senotherapeutic agent to mitigate cellular senescence observed in the case of vascular calcification.
Material and methods
Materials
Male Sprague Dawley rats (Charles River, CD® IGS strain code-001), Adenine diet containing 2.5% protein (Envigo Teklad Custom Diet TD. 130127, Madison, WI), EDTA (Sigma, E6511), vitamin D3 (VitD3, Cholecalciferol, Sigma, #C9756-5G), NaCl (sigma, S3014), KCl (Sigma P9541), NaHCO3 (Sigma, S5761), glucose (Sigma, G7021), NaH2PO4 (Sigma, S0876), dPBS (Gibco, 10010-031), DMEM (Gibco-1885-076), FBS, Pen-Sterp, Human Aortic Smooth Muscle Cells (SMCs) (Promocell, C12533), SMC Medium II (Promocell 22062), ABT263, HSA (Evonik Birmingham Laboratory, 777HSA097), HEPES (Sigma, H3375), absolute ethanol (Fisher BP 2818500), mPEGNHS, MW 2000 (BOC Sciences), Traut’s reagent (G-Biosciences, Saint Louis, MO, BC95), 5-Dodecanoylaminofluorescein di-β-D-Galactopyranoside (C12FDG, MedChem Express HY-126839), Cell Staining buffer (BioLegend 00-4222-26), anti-CD16/32 mouse monoclonal antibody (BioLegend Cat#156604), anti-CD45-APC (eBiosciences Cat#17-0461-82) DAPI (BioLegend Cat#422801), Rabbit anti-Rat Caspase-3 (Cell Signaling Technology, C8487), NLRP3 (Novus Biologicals, NBP2-12446), and PiT-1 (Santa Cruz Biotechnology, SC98824), anti-rabbit IgG secondary antibody Cy5 (Invitrogen, A10523) cDNA synthesis Kit (Bio-Rad, Cat# 1725034), iTaq—Universal SYBR Green Super mix 12 reagent (Bio-Rad, Cat# 172512), SA-βGal Staining Kit (Cell Signaling, 9860), Rat IL-6 ELISA Kit (RnD SYSTEMS DY-506), Rat IL-1β ELISA Kit (RnD SYSTEMS SRLB00), T-Per protein extraction buffer (ThermoFischer, 78510), BCA assay kit (Thermo Fisher Scientific, A55865).
CKD-induced vascular calcification model
To induce late-stage chronic kidney disease (CKD) and medial vascular calcification, animals were fed a 0.75% adenine diet (2.5% protein) for 28 days. After completion of the adenine diet, animals were transitioned to a standard chow diet for 5 days (days 29–33) to allow stabilization. From days 33 to 36, all adenine-treated animals received daily intraperitoneal injections of calcitriol (vitamin D3, 8.75 mg/kg/day in olive oil) to accelerate vascular calcification. Three days after the final vitamin D3 dose (day 39), animals were randomized into two CKD treatment groups; EDTA NP group received intravenous injections of EDTA-loaded nanoparticles (10 mg/kg) every 3 days for a total of five doses (days 39, 42, 45, 48, and 51); Blank NP group: received equivalent intravenous doses of non-chelating (blank) nanoparticles on the same schedule. In parallel, a group of healthy reference animals maintained on standard chow without adenine or vitamin D3 exposure was included for baseline comparison.
All animals were monitored daily, and humane endpoints were observed throughout the study. Animals exhibiting >20% body weight loss or signs of severe morbidity were euthanized before the planned endpoint (day 56). At euthanasia, blood was collected for serum biomarker analysis, and organs, including the aorta, kidneys, liver, lungs, and spleen, were harvested and preserved for histological and biochemical analysis. Notably, only 3 of the 12 CKD animals completed the full 5-dose nanoparticle treatment course; the remaining 9 were euthanized early due to humane endpoints (Supplemental Figure 1).
Aortic ring culture
Male Sprague-Dawley rats (16 weeks) were euthanized by saline perfusion under isoflurane anesthesia for harvesting full-length aortas. Aortas were collected in an aseptic condition in ice-cold Moscona’s buffer (8 g NaCl, 0.2 g KCl, 1 g NaHCO3, 1.7 g glucose, 0.005 g NaH2PO4 in 1 l DI water; pH 7.2) and transported to the lab. After washing with Dulbecco’s phosphate-buffered saline (PBS), the aortas from the ascending to the iliac bifurcation were cut into pieces to obtain 1 cm aortic rings. These rings were then cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 1% heat-inactivated FBS and 1% penicillin-streptomycin in a humidified incubator at 37°C, under 5%CO2 for overnight equilibration. The aortic rings were treated with 0.5 U/ml elastase for 40 min at 37°C to induce initial elastin damage. Subsequently, elastase-treated aortic rings were washed twice with PBS and incubated in high phosphate medium in DMEM (final concentration of Pi: 2.9 mM) containing 1% heat-inactivated FBS and 1% penicillin-streptomycin for ten days. The rings were then cultured with 0.5 mg/ml pure EDTA (corresponding to the same concentration as delivered in vivo with HSA nanoparticles) in the presence of high phosphate in DMEM and 10% FBS for 3 days. The experimental groups were: (i) DMEM only (0.9 mM phosphate; denoted as a control group); (ii) elastase & high phosphate treated with no EDTA (high Pi group); (iii) elastase & high phosphate treated followed by EDTA (EDTA-treated group). NaH2PO4.2H2O in a final concentration of 2.9 mM was used as a source of Pi. During incubation, the conditioned medium was replaced every 2 days (Supplemental Figure 2).
Cell culture studies
Human aortic smooth muscle cells (HAoSMCs) were purchased from Promocell and used between passages four and five for all experiments. Monocultures were maintained in 75 cm2 tissue culture-treated vented flasks in a 37°C and 5% CO2 environment in Smooth Muscle Cell Medium II (Promocell). To create high phosphate conditions, NaH2PO4 · 2H2O was added to obtain a final concentration of 2.9 mM of Pi. EDTA was added to achieve the final concentration of 0.5 mg/ml in the EDTA treatment group in the culture media. The final working concentration of ABT263 used in the study was 1 mM.
EDTA Np preparation and conjugation with Flexibzumab antibody
The desolvation method was used to make EDTA-loaded human serum albumin (HSA) nanoparticles following the process described earlier 19 with slight modifications. Briefly, 200 mg of HSA (SeraCare, Milford, M.A.) was dissolved in 4 ml of deionized water, and 50 mg of disodium salt of EDTA (Fisher Scientific, NJ) was dissolved in the HSA solution. The pH of the solution was adjusted to 8.5. The solution was then added dropwise to the absolute ethanol (1 ml/min) under constant stirring, followed by 25 µl of 8% glutaraldehyde as a crosslinker. The solution was incubated at room temperature for 2 h with continuous stirring at 800 rpm. Nanoparticles thus formed were centrifuged at 6000 rpm for 10 min, rinsed in deionized water (saturated with EDTA), and resuspended in phosphate-buffered saline before conjugating with thiolated anti-elastin antibody conjugation. 27 10 mg of formulated nanoparticles were PEGylated with 2.5 mg of α-maleimide-ω-N-hydroxysuccinimide ester poly (ethylene glycol) for an hour at room temperature under gentle agitation. Meanwhile, 20 µg of custom-made humanized anti-elastin antibody was added to 68 μg of Traut’s reagent for antibody thiolation. Subsequently, the mixture was incubated in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (20 mM, pH = 8.8) at room temperature for an hour under gentle agitation. Finally, the thiolated antibody was added to the PEGylated nanoparticles and incubated overnight (16 h) at 40°C under gentle rocking for conjugation.
SA-beta galactosidase activity
Cells isolated from the abdominal aorta were counted and adjusted to 3 × 106 cells/ml in complete culture media, sterile DMEM containing DNase, and incubated with 66 mM C12FDG for 1 h in a 37°C water bath. Cells were then spun down at 500 g for 5 min, washed with Cell Staining buffer (BioLegend), blocked with anti-CD16/32 mouse monoclonal antibody (BioLegend Cat#156604), and stained with anti-CD45-APC (eBiosciences Cat#17-0461-82) each at a 1:100 and 1:200 dilution on ice for 30 min. Cells were then washed in Cell Staining buffer and resuspended in Cell Staining buffer containing 1X DAPI (BioLegend Cat#422801). Samples were analyzed on an Attune acoustic focusing cytometer. Gates were set as follows: cells (FSC-A/SSC-A), forward scatter singlets (FSC-H/FSC-A), side-scatter singlets (SSC-H/SSC-A), live cells (DAPI-ve), and non-immune cells (CD45-ve), and were analyzed for C12FDG fluorescence. Data were analyzed with FlowJo (v.10). 28
Immuno-histochemistry
Abdominal aortas with cryosections of 7 µm thickness were fixed in cold acetone for 10 min, followed by rehydration along with decalcification in 0.5% of disodium ethylene diamine tetraacetic acid in Dulbecco’s phosphate-buffered saline (PBS) for 15 min at room temperature. The sections were then washed in PBS and subsequently blocked with Background Buster for 30 min at room temperature and incubated at 4°C overnight with primary antibodies: Rabbit anti-Rat Caspase-3 (Cell Signaling Technology, Danvers, M.A.), NLRP3, and PiT-1 (Santa Cruz Biotechnology, Dallas, Texas). Stained sections were then incubated with appropriate secondary antibodies conjugated with either anti-rabbit Cy3 or Cy5 IgG for visualizing the target proteins. Finally, the aortic sections were counterstained with DAPI and mounted with an aqueous mounting medium for imaging. Four to six sections from equal spacings were taken from each sample for the semiquantitative analysis of target proteins. ImageJ software was used for image analysis. Normalization was performed against DAPI-stained cells. The DAPI channel image was converted to an 8-bit gray scale, and the same threshold was applied to all the photos henceforth. The Analyze particle function was used to remove debris (particles between 30 um and 1000 um size range were selected), and the total number of DAPI-positive cells was counted in different fields. The image was opened in the target channel, and the entire field was selected as ROI. Then the analysis function was used to measure the integrated density (sum of pixel intensities × area) for the marker within the ROI. Normalization of signal was done using the following formula: Normalized Marker Intensity = Integrated density (Marker)/No. of DAPI +ve cells.
Reverse transcriptase quantitative PCR for gene expression
The expression of genes of interest was performed in aorta samples. The tissue was stored immediately after retrieval in the Trizol. The tissue was homogenized in Trizol using Powergen 125 (FS-PG125, Fisher Scientific) homogenizer, and RNA was extracted using the phenol-chloroform extraction method. A nanodrop instrument was used to analyze the extracted RNA through quantitative and qualitative analysis. cDNA synthesis was performed using 200 ng of total RNA with iScript gDNA clear cDNA synthesis Kit (Bio-Rad, Cat# 1725034). The total cDNA obtained was diluted five times before amplification with iTaq—Universal SYBR Green Supermix 12 reagent (Bio-Rad, Cat# 172512) on the Bio-Rad quantitative PCR platform (96-well format). Quantification was performed using the delta-delta CT method, using GAPDH as a housekeeping gene. A list of Primers used (OCN, RUNX2, IL-6, IL-1β, MCP-1, p21, p19, and GAPDH) in the study is given in Supplemental Table 1.
IL-6, IL-1β measurements by ELISA
Culture supernatants were centrifuged at 2000 × g for 10 min at 4°C to remove cellular debris. Cytokine concentrations in cell culture supernatants and animal serum were analyzed using ELISA kits (Invitrogen) according to the manufacturer’s protocol.
MMP activity analysis using zymography
MMPs activity in the serum was measured using in-gel zymography, with slight modifications. 27 Briefly, serum samples from the treatment group (containing 20 µg of total protein) were loaded on gelatin-impregnated SDS gels (ZY00105, Fisher Scientific). Coomassie blue staining was used to visualize the sites of proteolytic digestion.
Microcomputed tomography (micro-CT) imaging for calcification
Calcification or mineral deposits in rat aortas and kidneys were scanned using microCT (Bruker Skyscan 1176, Billerica, MA). Immediately after sacrifice, rat aortas were explanted and kept in cold PBS in 50 ml tubes for micro-CT scanning. Aortic scanning was performed using a 0.5 mm Aluminum filter at a voltage of 90 kV and a current of 278 µA, with 45 ms exposure time. Aorta specimens were scanned using a 360° rotation with a step of 0.7°. The reconstruction of x-ray back-projection images into cross sections was performed using Bruker’s NRecon Software, which uses modified Feldkamp’s algorithms. The range of attenuation coefficients was the same across all aortas to 10, acquiring a comparable result between blank Nps and EDTA-NPs groups. For comparative analysis, Bruker’s volume rendering CTVox software was used to represent reconstructed images as a 3D object, with the same settings over aortas or kidneys. Further, 3D morphometric analysis was conducted with CTAn software from Bruker.
Alizarin red staining
Optimal cutting temperature (OCT) compound–embedded frozen sections (7 µm each) were stained with 2% Alizarin Red solution (pH 4.1–4.3) for 1 min. The stained sections were then washed with two changes of distilled water for 10 min each and subsequently dehydrated in graded alcohol and xylene for mounting and bright field imaging with Keyence BZ-810. Semiquantitative analysis for percentage positive stain was performed with equally distributed four to six serial sections from each abdominal aortic sample.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 10. Results were analyzed by a one-way ANOVA test. The number of biological replicates (n) is indicated in the figure legend. Statistical comparisons were performed between groups of rats, and/or samples, using two-tailed t tests for two groups, or one-way ANOVA for three, with corrections for multiple testing. p < 0.05 was considered significant. Sample sizes (N = 5–6 per group) were based on preliminary data and prior published studies demonstrating similar effect sizes in models of vascular calcification and senescence.27,28 An a priori power analysis was performed using Graph PRISM software (version 10) to detect a minimum effect size of 1.5 with 80% power and p = 0.05 for one-way ANOVA and unpaired t-tests. This analysis indicated that a minimum of five animals per group was sufficient to detect statistically significant differences in calcification, senescence markers, and NLRP3 expression. Where applicable, all experiments were independently replicated to ensure reproducibility and statistical robustness.
Proteomics analysis
A 3 cm section of abdominal aorta was homogenized, and protein was isolated using T-Perm protein extraction buffer per the manufacturer’s protocol (Thermo Fischer). Protein concentrations were determined using the BCA assay kit (Thermo Fisher Scientific, Waltham, M.A., USA). Protein samples were normalized to 60 µg with MS-grade water, and proteins were reduced with 20 mM tris (2-carboxyethyl) phosphine (TCEP) by incubating at 50°C for 15 min. Proteins were brought to room temperature and then alkylated with 40 mM iodoacetamide (IAA) by incubating in the dark at room temperature for 30 min. Tryptic digestion was performed using suspension traps (S-trap mini, Portici, Fairport, NY, USA) following the manufacturer’s protocol. The reduced and alkylated proteins (60 µg) were acidified with 10:1 sample/12% phosphoric acid (v/v) and then diluted with 1:7 acidified sample/Binding Buffer (v/v). Proteins were loaded to S-traps in aliquots of 200 μl, centrifuged at 4000 g for 30 s, discarding the flow-through, washed six times with 200 μl Binding Buffer, discarding the flow-through, and centrifuged at 4000 g for 1 min. Trypsin protease was added at a 1:10 trypsin/sample protein (w/w) ratio, centrifuged at 1000 rpm for 10 s, and incubated in a dark water bath at 37°C for 13 h. Peptides were eluted from S-traps with 50 mM ammonium bicarbonate in water, centrifuged at 3000 rpm for 1 min, repeated elution with 0.1% formic acid in water, and then with 40% acetonitrile containing 0.1% formic acid in water, combining eluates in one 2 ml tube. Peptides were concentrated by evaporation under a nitrogen gas stream and reconstituted to a final protein concentration of 1.2 g/l in 95% water, 5% acetonitrile, 0.1% formic acid, containing 50 nM diluted Pierce™ Peptide Retention Time Calibration Mixture.
Protein digests were analyzed on an UltiMate™ 3000 UHPLC (Thermo Scientific) coupled to an Orbitrap Fusion™ Tribrid mass spectrometer (Thermo Scientific) equipped with an EASY-spray™ nano-flow source. Two microgram protein digests in 1 µl injections were loaded onto PepMap™ RSLC C18 NanoSpray column (2 µm, 100 Å, 75 × 50 cm). Peptides were separated using a solvent gradient with 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in 80% acetonitrile (mobile phase B) at a flow rate of 250 nl min−1. For peptide separation, the column was initially equilibrated at 4% B for 3 min, increased to 30% B at 90 min, increased to 55% B at 120 min, increased to 90% B at 130 min, held at 90% B until 134 min, and then decreased to 4% B at 135 min. The solvent gradient included a column flush method consisting of three rapid gradient flushes of 4% B–90% B, holding at each for 4 min, then re-equilibrating at 4% for 25 min. Peptides were ionized in positive ionization mode using 2.2 kV spray voltage, 2 Arb sweep gas flow, and 275°C ion transfer tube temperature. The MS1 scan (m/z 300–1500) was performed in an Orbitrap mass analyzer at 500,000 resolutions with a cycle time of 2 s. MS2 scans were collected for ions that passed the following filters: peptide monoisotopic peak determination, charge states 2–7, dynamic exclusion duration of 40 s for 10 ppm mass tolerance, minimum intensity of 1.9E4, and isotope exclusion. MS2 scans were acquired in the ion trap mass analyzer with an isolation window of 1.2 amu, following activation with collision-induced dissociation (CID) of 35% energy. Data were processed in Proteome Discoverer (Version 3.1.0.638, Thermo Fisher Scientific) with FDR confidence <0.01, using FASTA files for Ratus norvegicus: Rattus norvegicus (sp_tr_incl_isoforms TaxID=10116_and_subtaxonomies), downloaded Oct 20; 83181 sequences. Normalized data was used from Proteome Discoverer for analysis. Features with >50% missing values were removed, for the remaining missing values were estimated with BPCA. The data was auto-scaled.
Results
Targeted chelation therapy with EDTA NPs acts as senomorphics and not senolytic
Taking leads from our previous findings and having established that targeted chelation therapy decreases the expression of BMP2and RUNX2 in this study using the CKD model, we first validated that therapy with EDTA-NP decreases calcium deposition in the aorta and increases longevity in treated animals. We observed a significant decrease in the concentration of inflammatory cytokines, IL-6 and IL-1β, in the serum and an improvement in the survival rate of the animals treated with EL-EDTA-NPs (Figure 1).
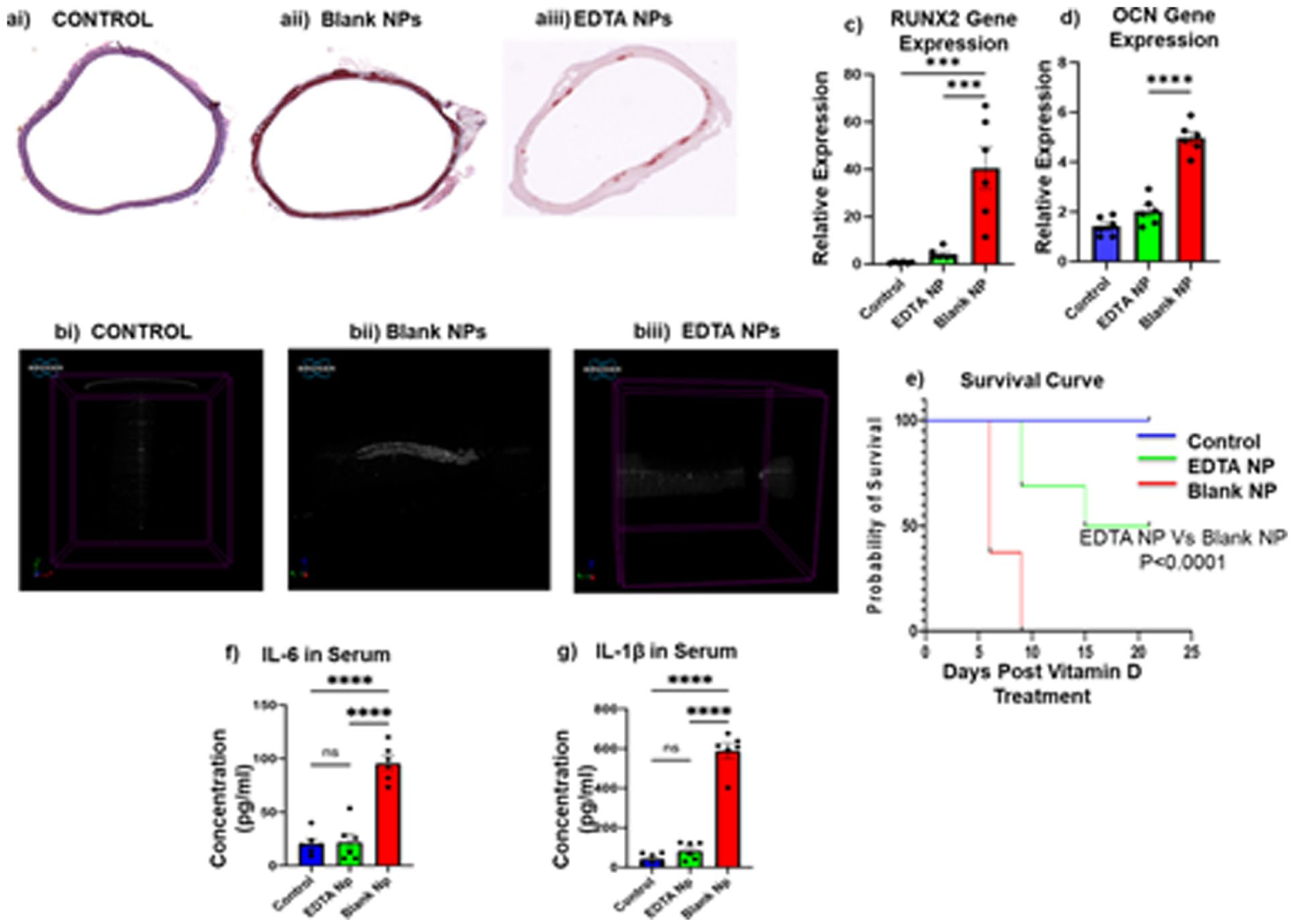
EDTA therapy increases Longevity in Late-Stage CKD rats by decreasing Calcification in the Aorta. Calcium deposition in the aorta was analyzed by using (a) Alizarin Red staining, and (b) Micro-CT scanning of the aortas. (c–d) Transcript level analysis of ossification markers RUNX2 and Osteocalcin (OCN). A decrease in their expression level demonstrates a decreased tendency toward osteoblastic phenotype switching. Further, we observed a significant improvement in the survival rate, as is reflected by the (e) Survival curve post vitamin D treatment with a simultaneous significant decrease in circulating levels of pro-inflammatory cytokine (f–g) IL-6 and IL-1β. N = 6 per group results represented as Mean ± SEM.
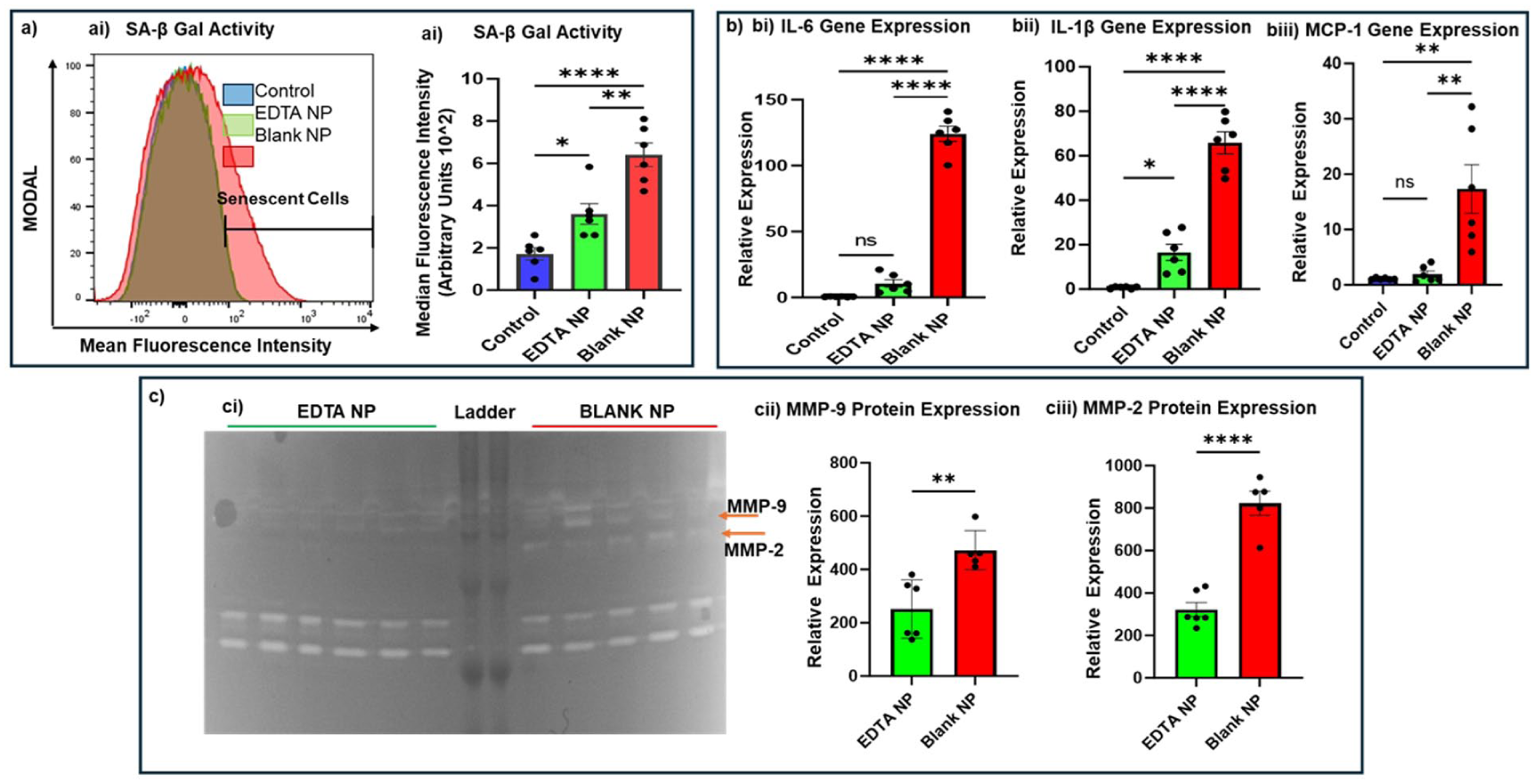
Senomorphic Effect of EDTA nanoparticles—Targeted therapy with EDTA NPs decreased the expression of SASP Markers significantly in aortas harvested from Late-Stage CKD animals (ai-ii) SA-β Gal Activity, (bi-biii) transcriptional expression of IL-6, IL-1β, and MCP1, as well as activity of (ci-iii) MMP 2 & 9. N = 6 per group, results represented as Mean ± SEM.
To examine whether EDTA induced apoptosis of senescent cells, we used a well-established senolytic agent ABT 263, which is known to induce apoptosis by recruiting NLRP3 via caspase3 activation and tested it against EDTA. We observed that EDTA treatment instead decreased Caspase 3 expression in primary human vascular smooth muscle cells exposed to high phosphate ion concentration, whereas ABT 263 treatment increased caspase-3 expression, indicating that EDTA does not induce NLRP3-mediated apoptosis. Furthermore, a decrease in Caspase 3 expression suggests transcriptional inhibition of NLRP3 expression during the priming stage (Figure 3(a) and (b)).

(a) EDTA is Senomorphic and not Senolytic—Immortalized human vascular smooth muscle cells cultured under high phosphate conditions for 3 days and were treated with either EDTA or ABT 263 (senolytic agent) for 24 h. IHC-based Expression level analysis of Caspase 3 was used as a marker for senolytic (inducing selective apoptosis of senescent cells) activity. We observed that in comparison to ABT 263, EDTA treatment significantly brought down Caspase 3 activity, indicating that EDTA does not induce apoptosis. Experiment performed in triplicate, repeated four times, results represented as Mean ± S.D and (b) EDTA but not ABT263 Decreases NLRP3 expression—Immortalized human vascular smooth muscle cells cultured under high phosphate conditions for 3 days and were treated with either EDTA or ABT 263 (senolytic agent) for 24 h. IHC-based Expression level analysis of NLRP3 reveals that EDTA treatment and not ABT 263 decreased NLRP3 activity. Justifying the anti-apoptotic and senomorphic nature of EDTA treatment. Experiment performed in triplicate, repeated four times, results represented as Mean ± S.D.
Targeted chelation therapy with EDTA NPs significantly decreased NLRP3 expression in calcified aorta
We examined the expression of NLRP3 in the calcified aorta harvested from late-stage CKD rats, as well as in the long-duration aortic ring culture model. In the calcified aortas harvested from both aortic ring culture and CKD model, we observed that treatment with EDTA and EDTA NPs, respectively, caused a significant decrease in NLRP3 expression as quantified using IHC, and qPCR as well as a decrease in the concentration of IL-1 β and IL-6 in the culture supernatants (Figure 4(a) and (b)).

Effect of EDTA NP treatment on NLRP3, IL-1beta, IL-6 and Caspase3 expression in (a–b) calcified aortas harvested from late-stage in vivo CKD model (a) separate cohort of N = 5 per group was set up, results represented as Mean ± SEM); (c-e) NLRP3 expression (ci-v); IL-1beta (d) and IL-6 (e) levels in calcified aortas and supernatant harvested after 10 days of ex vivo/Long-term in vitro culture under high phosphate conditions (N = 4 per group; experiment done in triplicate; results represented as Mean ± SD). EDTA treatment significantly decreased Caspase 3 and NLRP3 expression in the late stage in vivo CKD model of vascular calcification, as well as in the in vitro model.
Proteomics analysis reveals a significant decrease in SASP markers and a simultaneous increase in markers of angiogenesis—we performed an untargeted top-down proteomics analysis of total protein isolated from the abdominal aorta of EDTA NP treated versus Blank NPs treated animals
A total of 825 proteins were identified that were significantly differentially expressed between the two groups (Figure 5(a), Supplemental Table 2). The analysis revealed that 498 proteins were exclusively expressed in the Blank NP treated group and 200 proteins were exclusively expressed in the EDTA NP treatment group (Figure 5(b)). Amongst the top 25 upregulated proteins, 26% are directly influence cell proliferation and angiogenesis (Anaxa8, Enpep, Fgf8b, Clec11a, Crlf1, Agc1, Lancl2), 14% prevent osteoblastic transition of VSMCs (MGP, Phosphorylated form of Spp1, and ANKH), 14% play a role in cytoskeleton stabilization (RCG23467, Dmd, and Actb), 10% are involved in mediating proper protein folding (Ppidl1, Hmga1), 10% are involved in anabolic Lipid and carbohydrate metabolism (Mdh1 Mor2, Pla2g4a), and another 10% and 5% are anticoagulant and coagulants (Proc, and F7; Figure 5(c)).

Proteomic analysis of abdominal aorta from EDTA NP treated group and Blank NP treated group. (a) A volcano plot for significantly differentially expressed proteins—Upregulated (purple) or downregulated (red) in EDTA NP treatment versus the Blank NP treatment group. (b) Breakdown of total proteins identified during the analysis as exclusively expressed or differentially expressed in both the treatment groups to identify top hits. (c) Heat Map of differentially expressed proteins found in all the samples (di–dii) Breakdown of the top 25 upregulated and the top 20 downregulated proteins, respectively, as per biological functions. N ⩾ 5 per group.
Amongst the top 20 downregulated proteins, 35% were directly involved in facilitating SASP processes (Uchl1, H1-1 Hist1h1a, HP, Cfb C2, Tf, Serpina1), 15% were markers of vascular calcification (Ckmt2, Fetub, Pvalb Pva), 10% regulate muscle contraction (Myl2, RCG36716), 5% are involved in each Lipid metabolism, protein synthesis, and apoptosis (Iah1 Harpb64, Eef1a2 Kcnq2, Kng1; Figure 5(d)). We screened the list of downregulated proteins to scrutinize the ones that regulate NLRP3 activation. PIT-1, Csnk2b, Rbp4 regulates phase 1 of NLRP3 activation via Jak2/MapK/P13/AK. Protein S100a9 and Tf initiate phase 2 of NLRP3 activation and Jpt1 Sumo2, and PyCad are involved in stabilization of NLRP3 inflammasome complex. Amongst other downregulated proteins were the markers of senescence and SASP—Smap1, Serpina3k, Serpin2b, Bcl2l13, Bin1. To provide validation for the results, we evaluated the impact of EDTA on the expression of PIT-1, a critical determinant in NLRP3 activation. We observed that PIT-1 levels, which were significantly elevated by high phosphate levels, were brought down significantly by EDTA (Figure 6). This further strengthens our hypothesis that modulation of local mineral microenvironment by targeted chelation therapy can decrease SASP production in the tissue.
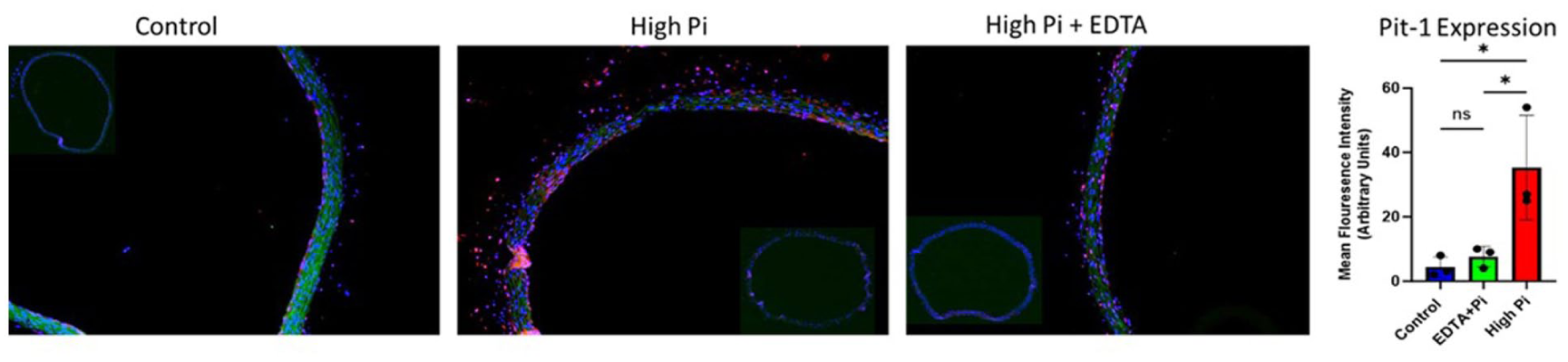
Effect of chelation therapy on PIT1—To validate the findings from the proteomics analysis, we used an in vitro ring culture method. Aortas harvested from the healthy animals were maintained in a high-phosphate environment for 10 days (we have previously published studies using this method). 39 IHC analysis reveals that EDTA treatment decreases PIT1 expression in the calcified aortas. The findings agree with our proteomics data.
Discussion
Senescent cells contribute significantly to the progression of various pathologies, including diabetes and chronic kidney disease (CKD), as well as to aging-associated vascular calcification. Even though senescent cells are in the stage of cell cycle arrest, these cells are metabolically active and possess a distinct inflammatory secretory phenotype that has the potential to modulate the tissue microenvironment. Senescent cells possess a distinct pro-inflammatory secretory phenotype known as SASP, which is responsible for the pervasive spread of senescence and in precipitating phenotypic transitions in the neighboring healthy cells within the tissue.28,29 In the context of vascular calcification in advanced CKD (stage 4 or 5), mineral imbalances—particularly elevated intracellular calcium and phosphate levels—play a pivotal role in driving phenotypic transitions within the tunica media.30–32 Clinical data from CKD patients reveal the accumulation of senescent cells and elevated SASP factors in the kidney, which coincide with the onset and progression of medial arterial calcification.9,33 A significant number of in vitro reports highlight the potential role of the inflammatory secretome from the senescent cell in osteoblastic differentiation of VSMCs. Despite promising preclinical findings, both the cellular origin of the SASP and the clinical translation of senolytic and senomorphic therapies for vascular calcification remain subjects of ongoing debate.8,34 One of the major challenges in advancing senolytics to the clinic is their off-target effects, including excessive apoptosis that can lead to hemorrhage and tissue damage. Notably, a recent study by Grosse et al. 35 demonstrated that global ablation of p16high senescent cells was not only non-beneficial but also detrimental, resulting in liver and cardiac fibrosis and compromising the integrity of the blood–tissue barrier, ultimately reducing health span and lifespan.
These findings underscore the critical need to further investigate the local sources of SASP and to develop targeted drug delivery strategies that can modulate the vascular microenvironment without inducing systemic toxicity. In this context, recent studies have identified NLRP3 inflammasome activation within calcified and inflamed cardiovascular cells as a key driver of SASP and a potential biomarker of cellular senescence in CKD-associated vascular calcification.
Our lab has also previously shown that culturing VSMCs under hyperphosphatemia conditions induces their transformation into osteoblast-like cells and upregulates the expression of biomarkers of cellular senescence, specifically BMP2 and RUMX2 in the VSMCs. 19 We have also previously established that targeted chelation therapy with EDTA NP can cause a reversal of vascular calcification and reverse the phenotype of ossified VSMCs in the aorta.19–21,27,36
In the present study, we investigated the serotherapeutic potential of EDTA chelation therapy in modulating vascular senescence. To assess cellular senescence, we employed a previously validated flow cytometry–based assay capable of detecting senescent cells in primary cells immediately after harvest from the tissue.27,28,37 This assay relies on the elevated activity of senescence-associated β-galactosidase (SA-βGal), a gold-standard marker of senescence. Using this standardized approach, we observed a significant reduction in the proportion of SA-βGal+ve cells in aortic tissues from EDTA NP treated animals compared to those treated with blank nanoparticles.
Next, we wanted to address whether the decrease in the senescent cell accumulation observed was due to senolytic (induction of selective apoptosis of senescent cells) property of EDTA. Senolytics are the agents that function by activating apoptosis pathways selectively in senescent cells. These often block the Anti-apoptotic BCL-2/BCL-xL signaling (e.g. Navitoclax), PI3K/AKT/mTOR survival pathways (e.g. Dasatinib), or p53/p21 checkpoint that are particularly upregulated in senescent cells and are used by senescent cells a as means to neutralize the pro-apoptotic pathways. ABT 263/Navitoclax specifically blocks BCL-2/BCL-xL signaling and frees up BAX/BAK to trigger MOPS and caspase 3 dependent apoptosis of senescent cells.
For this purpose, we compared EDTA with a well-established senolytic agent ABT263. ABT263 activates the Caspase 3 pathway by recruiting NLRP3 and induces apoptosis selectively in senescent cells. 38 In our study, we observed that, unlike ABT263, EDTA treatment decreased Caspase 3 and NLRP3 expression in the VSMCs maintained under high Pi conditions, whereas ABT263 had no impact either on NLRP3 activity or on the caspase 3 expression. Even though we expected ABT to show an increase in NLRP3 and Caspase 1 expression but a deviation from our expected results might have resulted because the IHC expression of these proteins was not normalized against cell count.
Next, we evaluated the effect of EDTA chelation therapy on the NLRP3, Caspase 1, and IL-1β expression in the abdominal aorta samples harvested from the CKD animal model treatment, and we observed a similar trend: a decrease in NLRP3, Caspase 1, and IL-1β expression in the EDTA treatment group versus the Blank NP treatment group.
Recent research documents the role of NLRP3 inflammasome activation as a source of SASP. Its activation has been linked to the precipitation of senescent phenotype in chondrocytes, osteoblasts, and adipocytes. NLRP3 activation has also been evidenced to promote arterial calcification by causing pyroptosis.39–41 Its activation leads to the conversion of pro-caspase 1 to caspase 1, ultimately inducing cell lysis. The overload of apoptotic debris produced in the process serves as the scaffold for Ca and P deposition and causes the remodeling of arterial tissue. In addition to its involvement in direct tissue remodeling, NLRP3 inflammasome activation also activates innate immune machinery in cells; it triggers the maturation of proinflammatory cytokines such as interleukin-1beta to engage innate immune defenses and hence plays a critical role in the progression of disease. In the context of chronic kidney disorder and aging-associated vascular calcification, experimental data show that the NLRP3 inflammasome pathway is activated in the kidney and VSMCs by klotho deficiency and doxorubicin (a well-known senescence inducer). 39
In this study, we observed that EDTA chelation therapy decreases PIT-1 expression in calcified aorta (Figure 6), and proteomic analysis revealed significant downregulation in S100A9 and Signal recognition particle receptor, both of which translate intracellular calcium concentration to activate NLRP3 inflammasome assembly. Furthermore, we observed a significant downregulation of PYD and CARD domain-containing peptide, which is primarily responsible for recruitment of proCaspase1, and its conversion to Caspase 1 by NLRP3 inflammasome complex. We also observed that the therapy decreased the expression of proteins that regulate osteogenic differentiation of VSMCs (Ckm2, FeutB, Vitamin D-binding protein) and increased the expression of proteins involved in angiogenesis (Enpep, Osteopontin and MGP) in the aortas harvested from the chronic kidney disease-based animal model of MAC. The animals also survived longer than the Blank NP treatment group, and we observed a significant decrease in SASP markers—Serpin1, Serpin3 3, BCL2, BIN. Our findings serve as proof of concept for using agents that can modulate ionic imbalance in the tissue microenvironment as senomorphics for targeting senescent cells to treat vascular stenosis by creating conditions suitable for vascular cells to reprogram their phenotype. This suggests that while senolytics remove senescent cells by hitting their Achilles’ heel—the very pathways that are upregulated in senescent cells to avoid death. This, however, may not be a suitable approach to target the cellular senescence in cardiovascular diseases, where sudden mass apoptosis induced by senolytics may lead to severe uncontrolled hemorrhage. By contrast, senomorphics like EDTA, which not only disarm senescent cells by suppressing their inflammatory output (e.g. via NLRP3, NF-κB, ROS) but at the same time also push them toward reprogramming (by downregulating BCL and caspase 3 and upregulating Enpep, Osteopontin and MGP) may prove to be a superior choice.
Conclusion
This study demonstrates that targeted EDTA-based chelation therapy not only reduces vascular calcification but also attenuates cellular senescence and NLRP3 inflammasome activation in the aorta under CKD conditions. By mitigating both calcium accumulation and inflammation-driven senescence, this strategy holds promise as a dual-action senomorphic and anti-calcific therapeutic approach. These findings support further exploration of nanoparticle-based chelation as a viable intervention to delay or reverse vascular aging in CKD.
Limitations
The current study was performed to evaluate the role of a high phosphate environment on initiating senescence phenotype in aortic tissue overall. The question remains unanswered: how does mineral imbalance turn on SASP intracellularly? Even though the upregulation of PiT-1 expression hints toward the involvement of Pi+ ions, experimental evidence is still required to establish a link. Another interesting aspect is to investigate the pathways responsible for senomorphic activity of EDTA. Further, we acknowledge that the study would have benefited from the use of a direct pharmacological inhibitor of NLRP3. Another important limitation of this study is the variability in treatment exposure among animals due to early euthanasia based on humane endpoints. Only 3 of 12 animals received the full course of 5 nanoparticle doses, while the remaining animals received only 1–4 doses prior to death or euthanasia. This disparity in treatment duration introduces challenges in interpreting therapeutic efficacy, especially for survival and tissue-level outcomes. Although preliminary findings suggest a trend toward improved survival in the EDTA NP group, these results should be interpreted with caution due to the limited sample size and differential dosing. Future studies with larger cohorts, stratification based on disease severity, and improved survival support will be necessary to more definitively evaluate the impact of treatment duration on therapeutic outcomes.
Supplemental Material
sj-docx-1-iji-10.1177_03946320251391142 – Supplemental material for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification
Supplemental material, sj-docx-1-iji-10.1177_03946320251391142 for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification by Shivani Arora, Gregory Halsey, Fatema Tuj Zohora, Alyssa Swiss and Narendra Vyavahare in International Journal of Immunopathology and Pharmacology
Supplemental Material
sj-jpeg-2-iji-10.1177_03946320251391142 – Supplemental material for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification
Supplemental material, sj-jpeg-2-iji-10.1177_03946320251391142 for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification by Shivani Arora, Gregory Halsey, Fatema Tuj Zohora, Alyssa Swiss and Narendra Vyavahare in International Journal of Immunopathology and Pharmacology
Supplemental Material
sj-jpg-3-iji-10.1177_03946320251391142 – Supplemental material for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification
Supplemental material, sj-jpg-3-iji-10.1177_03946320251391142 for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification by Shivani Arora, Gregory Halsey, Fatema Tuj Zohora, Alyssa Swiss and Narendra Vyavahare in International Journal of Immunopathology and Pharmacology
Supplemental Material
sj-jpg-4-iji-10.1177_03946320251391142 – Supplemental material for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification
Supplemental material, sj-jpg-4-iji-10.1177_03946320251391142 for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification by Shivani Arora, Gregory Halsey, Fatema Tuj Zohora, Alyssa Swiss and Narendra Vyavahare in International Journal of Immunopathology and Pharmacology
Supplemental Material
sj-jpg-5-iji-10.1177_03946320251391142 – Supplemental material for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification
Supplemental material, sj-jpg-5-iji-10.1177_03946320251391142 for Targeted chelation therapy decreases NLRP3 expression by vascular cells and acts as senomorphic in chronic kidney disorder induced vascular calcification by Shivani Arora, Gregory Halsey, Fatema Tuj Zohora, Alyssa Swiss and Narendra Vyavahare in International Journal of Immunopathology and Pharmacology
Footnotes
Acknowledgements
We would like to thank Dr. Agnes Nagy-Mehesz at S.C. Bio Craft COBRE Core and Dr. Nishanth Tharayil, as well as Elizabeth Leonard at the MUAL—Multi-User Analytical Lab and Metabolomics Core, at Clemson, for their help and support.
Author contributions
Narendra Vyavahare and Shivani Arora—Conceived the study, designed, and performed the experiments, drafted, revised, and edited the manuscript, Gregory Halsey and Fatema-Tuj Zohora, Alyssa Swiss—Executed the experiments, Narendra Vyavahare—Mentored the study design, revised the manuscript.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: NRV holds a significant Equity interest in Elastrin Theraputics Inc., which has licensed targeted chelation therapy with nanoparticles from Clemson University. However, this work is independently performed with NIH funding to NRV.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded partly by grant funding from the National Institutes of Health to NRV (R01HL133662, R01HL145064, P31GM131959).
Ethics approval
This study was approved by the Institutional Animal Care and Use Committee (IACUC) at Clemson University (Animal Use Protocol number 2021-006).
Data availability statement
All the relevant data are provided in the manuscript. The data that supports the findings of this study are available from the corresponding authors, [NRV and SA], upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.