
Abstract
Pyoderma gangrenosum (PG) is a rare noninfectious neutrophilic dermatosis characterized by recurrent, painful ulcers that commonly affect the lower extremities but can also involve other parts of the body. Over half of patients with PG have concomitant systemic immune diseases, with the association of PG with systemic sclerosis (SSc) being extremely rare. Treatment of PG primarily involves local therapy, steroids, and immunosuppressants, with an increasing emphasis on biologic agents. Among these, tumor necrosis factor-alpha (TNF-α) antagonists are considered effective. The patient in this report was an elderly female with a history of systemic sclerosis for many years and initially presented with gangrenous ulcers on the fingertips. After inconclusive conventional treatment, adalimumab was added for 5 weeks, resulting in disease suppression, a reduction in ulcer size, and re-epithelialization of the skin lesions after 6 months.
Introduction
Pyoderma gangrenosum (PG) is a rare neutrophilic dermatosis characterized by rapidly progressing, noninfectious, and painful ulcers with undermined edges and a violaceous halo. Due to its low incidence and lack of definitive laboratory or histopathological diagnostic criteria, PG is often misdiagnosed as an infectious disease, vasculitis, or malignancy. More than half of patients with PG have concomitant systemic autoimmune diseases; however, the coexistence of systemic sclerosis (SSc) and PG is exceptionally rare, with only a few case reports documented. 1 The primary treatment for PG involves local therapy, corticosteroids, and immunosuppressants. Recent studies increasingly support the use of biologics, particularly tumor necrosis factor-alpha (TNF-α) antagonists, in managing PG. 2 This paper presents a case of refractory PG in a patient with a long-standing history of SSc, where conventional therapies were ineffective, but the addition of adalimumab resulted in rapid symptom improvement.
Case report
The patient is a 60-year-old female with a history of systemic sclerosis (SSc), pulmonary arterial hypertension, and pulmonary interstitial fibrosis. She intermittently took prednisone acetate, hydroxychloroquine sulfate tablets, bosentan, and other treatments. The patient presented with skin thickening on the backs of both hands and forearms. Raynaud’s phenomenon is also positive. The patient has radiating striae around their mouth and tight facial skin. She presented with necrosis and ulcers on the fingertips of the third finger, which gradually progressed to multiple ulcers on the limbs and trunk. Despite debridement procedures, the affected area continued to expand, with the ulcers on the lower limbs being the most prominent, with dark red granulation tissue visible inside the ulcers, accompanied by a significant amount of purulent discharge with bloody exudate (Figure 1a). The patient experienced significant pain in the lower limb ulcer and required continuous use of oxycodone, with a visual analog scale (VAS) pain score of 5. The Rodnan score was 24 points.

(a) The patient presented with ulcers on both lower limbs, with deep ulcers with irregular edges on all four limbs. The edges of the lower limb ulcers are purplish-red, which is consistent with the clinical manifestations of pyoderma gangrenosum (PG). (b) Photos of the patient’s lower limbs at discharge. (c) Follow-up visit in June shows the condition of the patient’s lower limbs; as seen in the images, the ulcers have completely healed, forming scar changes resembling “cigarette paper.” (d) Gulliver’s sign in pyoderma gangrenosum. Note the new epithelial growth connecting the ulcer bed to the surrounding normal skin.
Upon admission, relevant investigations revealed a C-reactive protein (CRP) level of 65.52 mg/L and an erythrocyte sedimentation rate (ESR) of 66.0 mm/h; anti-Scl-70 antibodies: positive; absolute CD4+ T-cell count: 359.12/µL; echocardiography: pulmonary hypertension; chest CT: pulmonary interstitial fibrosis; and esophageal dilation. Secretion culture revealed Pseudomonas aeruginosa. The initial treatment considered SSc-related vasculitis with infection. The patient was initially treated with 20 mg of methylprednisolone daily for 10 days, followed by a taper to 8 mg orally once daily (QD). Additionally, the patient received 10 g of intravenous immunoglobulin (IVIG) for three consecutive days. Based on antibiotic susceptibility testing, amikacin, imipenem, and linezolid were sequentially administered. To improve circulation at the site of the ulcer, the patient was prescribed bosentan at a dose of 62.5 mg twice daily (BID), along with hyperbaric oxygen therapy. However, there was no significant improvement in symptoms. Skin pathology revealed neutrophil infiltration, inflammatory exudate, necrotic tissue, and some keratinization, along with epidermoid cysts (Figure 2a and b). The dermatology consultation resulted in a diagnosis of “pyoderma gangrenosum.” Following the consultation results, surgical debridement was immediately stopped, and the intensity of antibiotic therapy was reduced. The treatment plan continued with intravenous immunoglobulin and low-dose oral methylprednisolone. Adalimumab was added, starting with an initial dose of 80 mg followed by 40 mg weekly for 5 weeks. After continuous treatment, the CRP and ESR levels normalized. Upon follow-up in June, the leg ulcers had completely reepithelialized, leaving scars (Figure 1b and c).
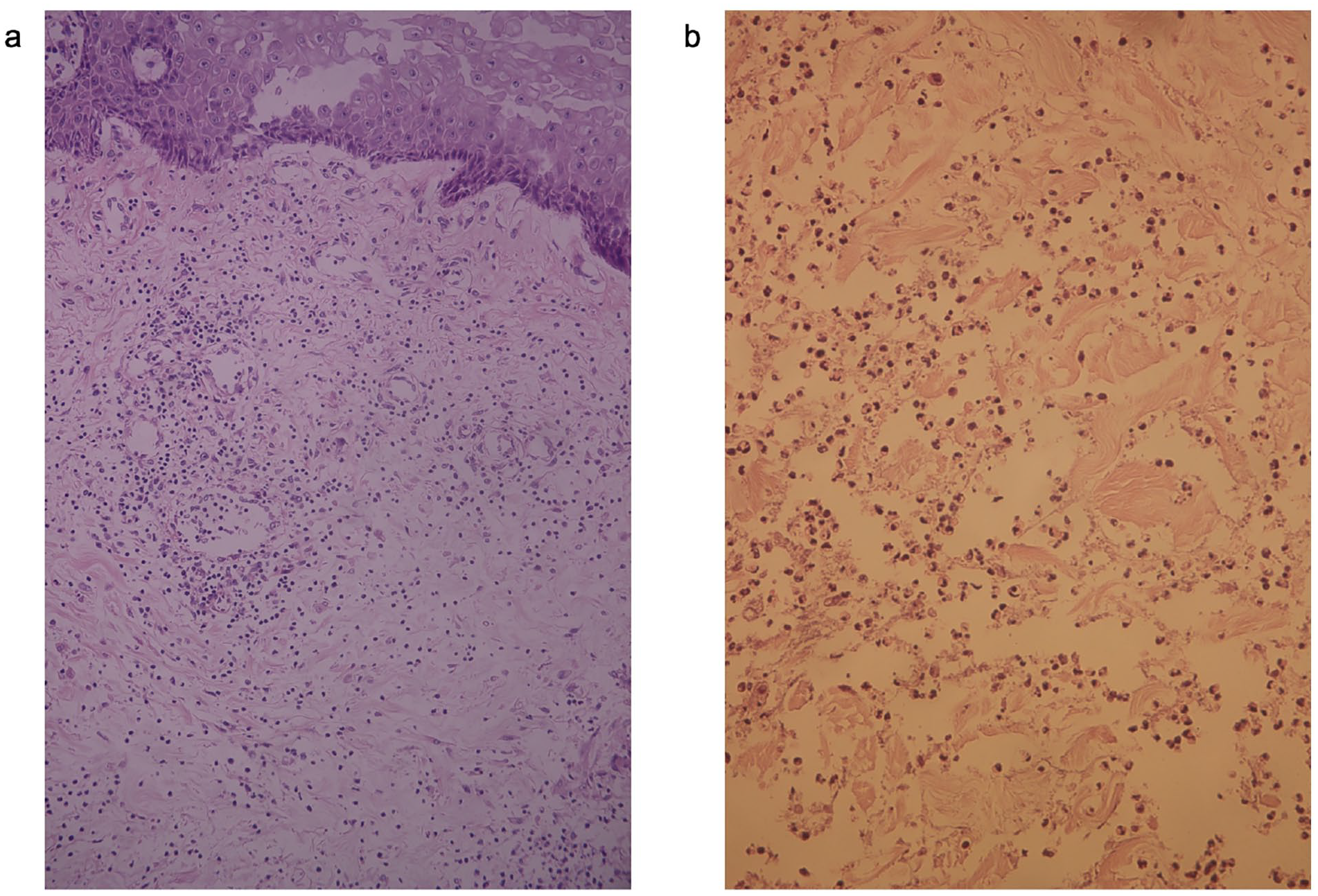
(a) ×20 magnification and (b) ×200 magnification skin biopsy of the lower limbs showing neutrophil infiltration, pyogenic dermatitis, and epidermal vesicles.
Discussion
Pyoderma gangrenosum (PG) is a rare and severe inflammatory skin condition with an annual incidence rate of 58 per 1,000,000 people. 3 Adults aged 40–60 years are most susceptible to PG, with a slightly higher prevalence in women than in men. 4 PG primarily manifests as recurrent, painful ulcers, most commonly on the lower extremities, although it can affect various parts of the body. Increasing evidence suggests that PG is a systemic autoinflammatory disease. 5 Patients with PG often have concurrent systemic inflammatory diseases, 6 which may also present as part of an autoinflammatory syndrome. A multicenter meta-analysis involving 2,611 patients 7 revealed that 56.8% of patients with PG also had other systemic diseases, including inflammatory bowel disease (IBD) in 17.6%, inflammatory arthritis in 12.8%, hematologic malignancies in 8.9%, and solid tumors in 7.4%. Additionally, the comorbidities associated with PG vary by ethnicity. Among Korean patients, hematologic malignancies are the most common comorbidity (24.5%), whereas in Japan, 30% of patients with large-vessel vasculitis are associated with PG.8,9
Systemic sclerosis (SSc) is an autoimmune disease characterized by localized or diffuse skin thickening and fibrosis. Patients with either PG or SSc can initially present with skin lesions. SSc-related skin ulcers often manifest on the fingers, wrists, or extensor surfaces of the elbows, typically with dry gangrene at the extremities. Vascular changes involve mainly proliferative small and medium-sized arteries, leading to luminal narrowing. In contrast, PG typically begins with painful erythema, papules, nodules, or pustules that rapidly progress into enlarging ulcers with characteristic purple-red borders and a necrotic base, sometimes extending into the muscle with associated suppuration and hemorrhage. 10 Histopathologically, PG is characterized by dense neutrophilic infiltration, although in chronic and recurrent cases, infiltration by histiocytes, lymphocytes, and fibrosis is more common. In this case, the patient presented with digital gangrene, rapidly progressive skin ulcers with irregular, undermined edges, severe pain, and a history of SSc. The PARACELSUS score for pyoderma gangrenosum was greater than 10, meeting the diagnostic criteria for PG. 11
The pathogenesis of PG may be related to neutrophil extracellular traps (NETs), a specific form of neutrophil death that releases proinflammatory and chemotactic factors, exacerbating local inflammation. 12 Damaged tissues release pathogen-associated molecular patterns (PAMPs), which, when recognized by immune cells, lead to cytokine secretion by neutrophils and macrophages, causing further tissue damage. The levels of the key proinflammatory cytokine TNF-α and its receptor are significantly increased during the inflammatory process of PG. 13
TNF-α inhibitors, including infliximab, adalimumab, and etanercept, are the most commonly used biologics in the clinical treatment of PG. A large study conducted in 2013 demonstrated that infliximab and adalimumab were more effective in treating PG than were corticosteroids, cyclosporine, or other immunomodulatory drugs. Among all therapies, TNF-α inhibitors provide the fastest therapeutic response and are well tolerated by patients. 14 According to the Japanese guidelines for PG, 2 adalimumab is recommended for patients with PG who do not respond well to steroids and cyclosporine. A study evaluating the treatment of refractory PG with adalimumab in Japan administered an initial dose of 160 mg, with 55% of participants achieving complete recovery at the end of the 26-week treatment period. 15 In this case, due to the high risk of infection, a reduced dosage was used following the treatment regimen in the literature. The patient’s wounds improved rapidly, with epithelial cells extending from the edges to the ulcers, showing “Gulliver’s sign” 16 (Figure 1d) and ultimately forming atrophic stellate scars.
Considering the patient’s history of interstitial lung fibrosis, the use of tumor necrosis factor (TNF) inhibitors poses a risk for acute lung injury. Most reported cases of adalimumab-induced lung injury occur between 2 and 6 months of treatment, although some cases with a shorter onset time have been reported. 17 Therefore, it is crucial to monitor the patient’s lung function continuously, be vigilant for symptoms of respiratory distress, and perform regular follow-up CT scans of the lungs. Infections are more common adverse effects of adalimumab. In a study conducted in Japan on adalimumab treatment for PG, 18 adverse events were reported, 11 of which were related to infections. Given that the patient has a low CD4+ count, has been bedridden for an extended period due to ulcer pain, and has been using steroids, they are considered immunosuppressed. Therefore, even after a definitive diagnosis, the patient continues to receive immunoglobulin therapy. This approach helps reduce the risk of infection while blocking the antibody and suppressing immune inflammation.
Previous case reports of SSc combined with PG have shown that the patients had good responses to moderate doses of hormones and immunosuppressants. In this case, the patient had extensive skin ulceration and high levels of inflammatory markers, and conventional doses of hormones and other traditional treatments did not improve her symptoms. Therefore, a more potent anti-inflammatory treatment is needed. Despite hormones being a first-line treatment for PG, the use of hormones in SSc treatment is relatively cautious. The risk of accelerated skin hardening and triggering of SSc renal crisis with long-term, high-dose use of glucocorticoids is a concern. Therefore, biologics are particularly suitable for patients in this category. biological agents are gradually playing a more important role in the treatment of PG. Increasing evidence in recent years suggests that biological agents are effective for PG. In addition to TNF-α inhibitors, case reports have shown the efficacy of IL-1 inhibitors, IL-17 inhibitors, IL-12/23 inhibitors, rituximab, and small molecule inhibitors in the treatment of PG.18–20 This provides more therapeutic options for patients with PG combined with rare systemic diseases, allowing for the reduction of adverse drug reactions.
Conclusion
Pyoderma gangrenosum (PG) has a low incidence, and currently, there is no standardized scoring system to assess its severity. Although more than half of PG patients have concomitant systemic diseases, the occurrence of PG combined with systemic sclerosis (SSc) is extremely rare. Both conditions primarily manifest as skin lesions, and PG lacks characteristic serological markers and pathological features, necessitating thorough differential diagnosis for a definitive diagnosis. The traditional combination of corticosteroids and immunosuppressants remains the mainstay of PG treatment. In previous case reports of PG combined with SSc, corticosteroids combined with cyclosporine, azathioprine, or other treatments have been employed, yielding satisfactory results. 17 However, considering the risks of long-term corticosteroid use in patients with SSc and the poor response of some PG patients to traditional treatment regimens, biological agents are gradually playing a more important role in the treatment of PG. This provides more therapeutic options for patients with PG combined with rare systemic diseases, allowing for the reduction of adverse drug reactions.
Footnotes
Author contributions
The case was diagnosed and followed by Yueyue Zhao and Maoquan Zhang. Chengqiang Ren conducted the literature search and analysis and completed the writing. Maoquan Zhang and Cheng Yu provided guidance on summarizing, synthesizing, and writing the medical records. The final version was read and proofread by Ding Li and Maoquan Zhang.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Informed consent
Written informed consent was obtained from legally authorized representatives before the study.
Trial registration
Not applicable.