
Abstract
Objectives
This experimental study aims to investigate the role of long noncoding RNA X-inactive specific transcript (lncRNA XIST) in the microglial polarization and microglia-mediated neurotoxicity in Alzheimer’s disease (AD).
Methods
The levels of XIST and microRNA-107 (miR-107) were detected by quantitative real-time polymerase chain reaction. The spatial learning and memory capability of APPswe/PS1dE9 (APP/PS1) mice were evaluated by the Morris water maze test. The morphology of mouse hippocampus cells was evaluated by hematoxylin and eosin staining. The Iba1-positive microglia were labeled by immunohistochemistry staining. The protein levels were determined by western blot and enzyme-linked immunosorbent assay. Neurotoxicity was evaluated by the terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling, caspase-3 activity, and Cell Counting Kit-8 assay. The XIST, miR-107, and AD targets were predicted by bioinformatics analysis.
Results
The level of XIST was increased in APP/PS1 mice, and XIST silencing ameliorated AD progression. XIST silencing suppressed microglia activation, microglial M1 polarization, and proinflammatory factor levels, but promoted microglial M2 polarization in APP/PS1 mice and Aβ1-42-treated BV-2 cells. XIST knockdown reduced Aβ1-42-induced microglia-mediated apoptosis and enhanced cell viability in HT22 cells. XIST silencing down-regulated miR-107 level and attenuated Aβ1-42-caused suppression of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling. Those effects of XIST silencing were attenuated by miR-107 inhibitor or LY294002.
Conclusion
Downregulation of XIST lessened Aβ1-42-induced microglia-mediated neurotoxicity by modulating microglial M1/M2 polarization, which may be mediated by the miR-107/PI3K/Akt pathway.
Keywords
Introduction
Alzheimer’s disease (AD) is currently the primary cause of dementia, and the number of deaths associated with it is increasing annually.1,2 In present clinical management, Managing AD in clinical settings presents a significant challenge to health status and medical security of elderly populations worldwide. 3 Symptoms of AD include cognitive deficiency and memory loss, as well as pathological features such as β-amyloid (Aβ) aggregation and neurofibrillary tangles. 4 Additionally, the presence of high levels of Aβ peptide in the brain can cause microglial infiltration during AD development. Microglia cells that are activated by Aβ promote its clearance via the TREM2 surface receptor. Meanwhile, these activated microglia cells trigger an immune response to inhibit the aggregation of Aβ.5,6 Neuroinflammation is characterized by the activation of immune cells, such as microglia and astrocytes, in the brain and nervous system. These cells release various cytokines and inflammatory mediators, such as interleukins, tumor necrosis factor, and interferons, which can lead to neuronal damage and inflammation, thereby exacerbating the symptoms of AD. 7 Unfortunately, the current therapeutic options for of AD are very limited, and targeted therapy is still an area of ongoing research. 8 Thus, there is an urgent need to identify effective targets for the management of AD.
Long noncoding RNA (lncRNA) is RNA that is longer than 200 nucleotides and does not encode protein. 9 Numerous studies have shown that lncRNAs play a crucial role in gene regulation and cellular functions during neuroinflammation in central nervous system disorders, including AD.10,11 Several lncRNAs, such as BACE1-AS, EBF3-AS, and EBF3-AS, have been demonstrated to be involved in AD development and have been identified as potential biomarkers for AD management.12–14 Besides, Du et al. have confirmed that lncRNA X-inactive specific transcript (lncRNA XIST) is upregulated in H2O2-induced N2a cells, which serves as a model of AD). Knockdown of XIST has been found to suppress AD by acting as a competitive endogenous RNA (ceRNA) via sponging miR-124 to regulate gene expression. 15 CeRNAs are a group of RNAs that can compete for miRNA binding with other target RNAs, such as mRNAs, thereby preventing their degradation or translation inhibition by miRNAs. Our previous study has shown that knockdown of XIST can reduce Aβ25-35-induced toxicity, oxidative stress, and apoptosis in primary cultured rat hippocampal neurons by targeting miR-132. 16 However, the effects of XIST on microglial M1/M2 polarization and neurotoxicity in AD are still unclear. Therefore, in this research, exhaustive studies will be performed. MiRNA is involved in various biological processes of cells, including proliferation, polarization, inflammatory response, differentiation, and apoptosis.17–20 Moreover, miRNA is an important regulator of AD. For example, miR-29a/b-1 can regulate the abundance of BACE1 in AD. 21 The study by Zhou et al. has revealed that the abundance of miR-383-5p in the plasma of AD patients is significantly lower, and miR-383-5p may serve as a diagnostic marker for AD. 22 Furthermore, the content of miR-107 was reduced in AD and accelerates disease progression. 23 However, the detailed effects of miR-107 in AD are still unclear. In addition, the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway is a key player in the inflammatory reactions of AD. 24
Herein, the APP/PS1 mice and Aβ1-42-treated BV-2 cells were utilized as the models of AD. The goal of this research was to investigate the influence of XIST on the development of AD and the corresponding regulatory mechanism. These findings will help to elucidate the pathogenesis of AD.
Materials and methods
Animals and treatment
This experimental study aimed to explore the role of lncRNA XIST in the microglial polarization and microglia-mediated neurotoxicity in both in vivo and in vitro models of AD. The male 12-month-old APP/PS1 transgenic mice and wild type (WT) C57BL/6 littermates were obtained from the Model Animal Research Center of Nanjing University. The mouse short hairpin RNA (shRNA) targeting lncRNA XIST (sh-XIST) packed with lentiviral particles and negative control (sh-NC) were obtained from Genechem (Shanghai, China). The sh-XIST or sh-NC was gradually inoculated into the bilateral hippocampus of APP/PS1 mice, and named APP/PS1 + sh-XIST group and APP/PS1 + sh-NC group (6 per group), respectively. The behavior test was carried out 30 days after the injection. Finally, the mice were euthanized for subsequent other experiments. The animal study followed the Guide for the Care and Use Laboratory Animals of Henan Mental Hospital and complied with Regulations of the People’s Republic of China for the Administration of laboratory animals. All animal tests were permitted by the Ethics Committee of Henan Mental Hospital (ethical approval number: 2020–1011).
Quantitative real-time polymerase chain reaction (qRT-PCR)
The RNA was isolated from the hippocampus of mice or cells by using the Trizol reagent (TaKaRa, Tokyo, Japan). Complementary DNA synthesis was carried out by employing the PrimeScript real-time reagent kit (Takara) and MicroRNA TaqMan® RT kit (Applied Biosystems, Carlsbad, CA, USA) using RNA. The qRT-PCR was conducted with the SYBR Green Master Mix (Takara) on an ABI 7900 system (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and U6 were used as controls. The gene level was calculated by the 2-△△Ct method. The sequences of primers were as follows: lncRNA XIST forward, 5′-ACGCTGCATGTGTCCTTAG-3′ and reverse, 5′-GAGCCTCTTATAAGCTGTTTG-3’; miR-107 forward, 5′-GCCAAGCCCACTCAGCTGCCAGCC-3′ and reverse, 5′-GGCTGGCAGCTGAGTGGGCTTGGC-3’; U6 forward, 5′- CTCGCTTCGGCAGCACATA -3′ and reverse, 5′- AACGATTCACGAATTTGCGT -3’; GAPDH forward, 5′-TCTCTGCTCCTCCCTGTTCC-3′ and reverse, 5′-TTTTGTCTACGGGACGAGGC-3’.
Morris water maze (MWM) test
The learning and memory capabilities of mice were evaluated by using the MWM test. In brief, a round water pool was grouped into four equivalent areas, and a platform was positioned in the middle of one of the four areas. Mice were subjected to four training examinations with a maximum of 120 s to discover the platform every day for 7 days. The mice were positioned at a random location and discharged to discover the invisible platform. The test was performed at 24 h after the last trail, and the escape latency (time to arrive the platform) and the number of times crossing the platform were recorded using a tracking system (Noldus, Beijing, China).
Hematoxylin and eosin (HE) staining
HE staining was carried out based on a previous research. 25 In short, the brain samples were fixed and cut into 5 μm sections, dewaxed, stained with hematoxylin (Solarbio, Beijing, China), dehydrated, stained with eosin (Solarbio), washed, and transparentized. The morphology of mouse hippocampus cells was observed and captured under a light microscope (Nikon, Tokyo, Japan).
Terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) assay
TUNEL assay was implemented to measure cell apoptosis. In brief, the brain tissues were exposed to paraformaldehyde (4%; Solarbio) and embedded in paraffin (Solarbio). Then, the samples were cut into 5 μm sections. Paraffin embedded sections were dewaxed and hydrated by graded alcohols. For in vitro cells, cell slides were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 for 5 min and blocked in 3% H2O2 for 10 min. Subsequently, these sections and cell slides were stained by TUNEL reaction mixture for 1 h at 37°C (Solarbio). The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific, Waltham, MA, USA). Finally, the sections and slides were assessed through a fluorescence microscope (Nikon). Cell apoptosis was quantitatively analyzed based on percentage of TUNEL-positive cells/total nuclei.
Enzyme-linked immunosorbent assay (ELISA)
The Aβ1-42 level in brain tissues of mice and the levels of tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-1β were detected by ELISA utilizing mouse Aβ1-42 ELISA Kit (CUSABIO, Wuhan, China), mouse TNF-α ELISA Kit (Abcam, Cambridge, MA, USA), mouse IL-6 ELISA Kit (Abcam), and mouse IL-1β ELISA Kit (Abcam). For brain tissues, homogenates were centrifuged at 5000 g for 5 min at 4°C, and then supernatant was collected for subsequent assay. For in vitro cells, cell supernatant samples were centrifuged at 500 g for 5 min and the supernatant was collected for subsequent assay. The ELISA was performed in line with the protocols of the manufacturer. The biotinylated antibody (100 μL) was added to each well and incubated at 25°C for 60 min. The horseradish peroxidase (100 μL) was added to each well and incubated away from light at 25°C for 20 min. Termination solution (50 μL) was added to each well, and then the optical density value at 450 nm of each well was detected by a microplate reader (Thermo Fisher Scientific).
Immunohistochemistry (IHC) staining
The brain were fixed in embedding agent (Solarbio) and cut into 5 μm sections. The sections were exposed to the immunized blocking solution (Solarbio) for 1 h. After blocking, the sections were incubated with the primary Iba1 antibody (1:200; Abcam) at 4°C overnight. Then, the sections were washed by PBS and incubated with the fluorescent secondary antibody (CWBIO, Beijing, China) at 25°C for 1 h. Next, the nuclei were stained with DAPI (Thermo Fisher Scientific). Finally, the sections were sealed with anti-quenching seals (Solarbio) and stored at 4°C. The sections were observed under a confocal fluorescence microscope (Nikon).
Western blot
The tissues or cells were lysed using a RIPA buffer (Solarbio) and the amount of protein was assessed using a BCA kit (Solarbio). Subsequently, the protein was electrophoresed on a 12% SDS-PAGE gel and moved to a PVDF membrane (Solarbio). After blocking, the membrane was coped with primary antibodies overnight at 4°C. The antibody used were as follows: anti-CD32 (1:1000; Abcam), anti-iNOS (1:1000; Abcam), anti-Arg-1 (1:1000; Abcam), anti-CD206 (Invitrogen, Carlsbad, CA, USA), anti-p-PI3K (1:1000; Cell Signaling Technology, Boston, MA, USA), anti-PI3K (1:1000; Cell Signaling Technology), anti-p-Akt (1:1000; Abcam), anti-Akt (1:1000; Abcam), and anti-β-actin (1:1000; Affinity Biosciences, Changzhou, China). Finally, the membrane was treated with goat anti-rabbit IgG (1:2500; Abcam) at 25°C for 1 h. The protein blots were visualized using an ECL-Plus reagent (Invitrogen, Carlsbad, CA, USA) and captured by a chemiluminescence imaging system (Bio-Rad, Hercules, CA, USA). The protein bands were quantitatively analyzed using the Image J software (National Institutes of Health, Bethesda, MD, USA).
Cell culture, transfection, and treatment
The mouse microglial cell line BV2 and mouse hippocampal neuronal cell line HT22 were obtained from Procell Life Science & Technology Co.,Ltd. (Wuhan, China) and cultivated in their specific medium (Procell) comprising 10% fetal bovine serum in an moist incubator containing 5% CO2 at 37°C. The siRNA targeting XIST (si-XIST) and the siRNA control (si-NC); miR-107 inhibitor (anti-miR-107), control (anti-NC), and LY294002 (a PI3K/Akt signaling inhibitor) were obtained from Genechem. Lipofectamine 2000 (Solarbio) was used to implement cell transfection according to the manufacturer’s instructions.
For preparation of Aβ1–42 oligomers, Aβ1–42 was dissolved in 100% hexafluoroisopropanol into 1 mM solution, and then allowed to evaporate at 25°C for 2 h to remove residual hexafluoroisopropanol. The peptide film was dissolved into 1 mM solution with DMSO. This solution was diluted to 100 μM solution in PBS and incubated for 24 h at 4°C. To mimic the Aβ1–42-induced microglia-mediated neurotoxicity in AD, the BV2 cells were transfected with si-NC or si-XIST and then stimulated with Aβ1–42 (10 μM) for 24 h, and afterward the mediums were collected as conditioned mediums. The conditioned mediums were used to incubate HT22 cells for 48 h. To inhibit the PI3K/Akt signaling pathway, BV2 cells were coped with LY294002 (10 μM; Genechem) for 1 h before transfection.
Detection of caspase-3 activity
HT22 cells were seeded into a 6-well plate and then cultured for 48 h in the conditioned medium from BV2 cells with or without Aβ1–42 (10 μM) stimulation. The Caspase-3 activity Kit (Nanjing Jiancheng Bioengineering Research Institute, Nanjing, China) was exploited to detect caspase-3 activity of HT22 cells. The protein was extracted from cells and moved to 96-well plates with 50 μL reaction buffer and 5 μL caspase-3 substrate. After 4 h incubation, caspase-3 activity was assessed through a microplate reader (Thermo Fisher Scientific) at 405 nm.
Cell counting kit-8 (CCK-8) assay
HT22 cells were seeded into a 96-well plate at a density of 4000 cells/well and then cultured for 48 h in the conditioned medium from BV2 cells with or without Aβ1–42 (10 μM) stimulation. Next, the CCK-8 solution (10 μL; Solarbio) was supplemented to each well. After 1 h, the absorbance (450 nm) was assessed through employing a microplate reader (Thermo Fisher Scientific).
Bioinformatics analysis
The miRNA targets of XIST were forecasted by miRcode (http://www.mircode.org/) 26 and starbase (http://starbase.sysu.edu.cn/). 27 The AD targets were forecasted by GeneCards (http://www.genecards.org/). 28 The gene targets of miR-107 were forecasted by MicroT-CDS (http://www.microrna.gr/microT) 29 and starbase. The targets were used for KEGG pathway enrichment analysis through DAVID (http://david.abcc.ncifcrf.gov/). 30
Statistical assay
The data were displayed as the means ± standard deviation. Statistical analysis was performed by one-way analysis of variance (ANOVA) using the GraphPad Prism software (San Diego, CA, USA). A p value less than .05 was considered to be statistically significant.
Results
XIST silencing attenuates AD progression in APP/PS1 mice
Firstly, we detected the level of XIST in the hippocampus of mice. The expression level of XIST was increased in the hippocampus of mice in the APP/PS1 group compared with the WT group. Compared with the APP/PS1 + sh-NC group, the XIST level in the hippocampus of mice in the APP/PS1 + sh-XIST group decreased obviously (Figure 1(a)). As the XIST level was increased in the APP/PS1 mice, we speculated that XIST silencing might restrain the progression of AD. Therefore, the spatial learning and memory capability of mice were evaluated by MWM. The escape latency was significantly elevated in the APP/PS1 group compared with the WT group. However, the escape latency was reduced by XIST knockdown (Figure 1(b)). In comparison with WT mice, APP/PS1 mice had a lesser number of times crossing the platform. But, XIST silencing significantly increased the number of times crossing the platform in APP/PS1 mice (Figure 1(c)). Next, we assessed the morphology of mice hippocampus cells by HE staining. In WT mice, the neurons in the hippocampus were stained uniformly, the structure was normal, and the cell membrane and nucleus were obvious. In APP/PS1 mice, the hippocampal tissue structure was abnormal, the cell morphology was changed, the neuron nuclei were pyknotic, the cytoplasm was hyperchromatic, and the nucleus boundary was unclear. Compared with the APP/PS1 + sh-NC group, the morphology of hippocampal cells in the APP/PS1 + sh-XIST group was improved, with round nuclei and obvious nucleoli (Figure 1(d)). Results of TUNEL experiments showed that apoptotic neurons are labeled with green fluorescence (Figure 1(d)). Then, we inspected the influence of XIST on cell apoptosis and content of Aβ1-42. Compared with WT mice, the hippocampus of APP/PS1 mice displayed an increased number of apoptotic cells (Figure 1(e)) and Aβ1-42 level (Figure 1(f)). Compared with APP/PS1 + sh-NC mice, downregulation of XIST diminished cell apoptosis (Figure 1(e)) and the level of Aβ1-42 (Figure 1(f)) in the hippocampus of APP/PS1 mice. There was no statistical difference between the APP/PS1 group and the APP/PS1 + sh-NC group. Hence, we confirmed that knockdown of XIST could inhibit AD progression in APP/PS1 mice. XIST silencing attenuates AD progression in APP/PS1 mice. (a) The level of XIST in the hippocampus of mice was quantified by qRT-PCR. The escape latency (b) and the number of times crossing the platform (c) of mice were evaluated by MWM test. (d) The morphology of mouse hippocampus cells was assessed by HE staining. The cell apoptosis was measured by TUNEL assay. Scale bar: 50 μM. (e) Apoptosis was quantified by calculating the percentage of TUNEL-positive cells. (f) The level of Aβ1-42 in the hippocampus of mice was determined by ELISA. *p < .05 and ***p < .001. n = 6 mice in each group.
XIST silencing shifts microglial M1/M2 polarization in APP/PS1 mice
Then, we explored the influence of XIST on microglial polarization in APP/PS1 mice. Iba1 is a marker of microglia activation.
31
We labeled Iba1-positive microglia by IHC staining (Figure 2(a)). As displayed in Figure 2(b), the relative fluorescence intensity of Iba1 was enhanced in the hippocampus of mice in the APP/PS1 group compared with the WT group. Compared with the APP/PS1 + sh-NC group, the intensity of Iba1 in the hippocampus of mice in the APP/PS1 + sh-XIST group significantly weakened. CD32 and iNOS are markers of microglial M1 polarization, which is a proinflammatory phenotype. Arg-1 and CD206 are markers of microglial M2 polarization, which is an anti-inflammatory phenotype.
32
The protein levels of CD32 and iNOS (Figures 2(c)–(e)) were increased and Arg-1 and CD206 levels (Figure 2(c), (f), (g)) were decreased in the hippocampus of mice in the APP/PS1 group compared with the WT group. After XIST knockdown, the CD32 and iNOS levels (Figures 2(c)–(e)) were reduced and Arg-1 and CD206 levels (Figure 2(c), (f), and (g)) were elevated in the APP/PS1 + sh-XIST group compared with the APP/PS1 + sh-NC group. There was no statistical difference between the APP/PS1 group and the APP/PS1 + sh-NC group. These consequences indicated that XIST silencing reduced microglia activation and microglial M1 polarization, but contributed to microglial M2 polarization in APP/PS1 mice. XIST silencing shifts microglial M1/M2 polarization in APP/PS1 mice. (a) The Iba1-positive microglia were measured by IHC staining. Scale bar: 50 μM. (b) Quantitative analysis results of relative fluorescence intensity of Iba1. (c) Representative images of western blot were shown. (d–g) The quantified results of CD32, iNOS, Arg-1, and CD206 protein expression were shown. ***p < .001. n = 6 mice in each group.
XIST knockdown shifts microglial M1/M2 polarization in Aβ1-42-treated microglial cells
To confirm the effect of XIST on microglial polarization in Aβ-induced microglial cells, we divided BV2 cells into control, Aβ1-42, Aβ1-42+si-NC, and Aβ1-42+si-XIST groups. The BV2 cells in the control group were cultured normally. The BV2 cells in the Aβ1-42 group were stimulated with 10 μM of Aβ1-42 for 24 h. The BV2 cells in the Aβ1-42+si-NC or Aβ1-42+si-XIST group were transfected with si-NC or si-XIST, and then stimulated with Aβ1-42 (10 μM) for 24 h. Results showed that the level of XIST was increased in the Aβ1-42 group compared with the control group, while this influence was significantly attenuated after si-XIST transfection (Figure 3(a)). Besides, the CD32 and iNOS levels (Figures 3(b)–(d)) were significantly increased and the Arg-1 and CD206 contents (Figure 3(b), (e), and (f)) were decreased in BV-2 cells after Aβ1-42 treatment compared with the control group, whereas this influence was attenuated by XIST knockdown. As shown in Figures 3(g)–(i), the exposure of BV-2 cells to Aβ1–42 alone led to a significant increase in the levels of TNF-α, IL-6, and IL-1β. However, the levels of these proinflammatory cytokines were reduced by silencing of XIST in Aβ1–42-treated BV-2 cells. There was no statistical difference between the Aβ1-42 group and the Aβ1-42+si-NC group. Therefore, we concluded that XIST knockdown suppressed microglial M1 polarization, and reduced proinflammatory factor levels, while facilitated microglial M2 polarization in Aβ1–42-treated BV-2 cells. XIST regulates microglial M1/M2 polarization in Aβ-treated microglial cells. (a) The level of XIST in BV2 cells was detected by qRT-PCR. (b–f) The protein levels of CD32, iNOS, Arg-1, and CD206 were examined by western blot. (g–i) The levels of TNF-α, IL-6, and IL-1β were detected by ELISA. **p < .01 and ***p < .001. n = 6 mice in each group.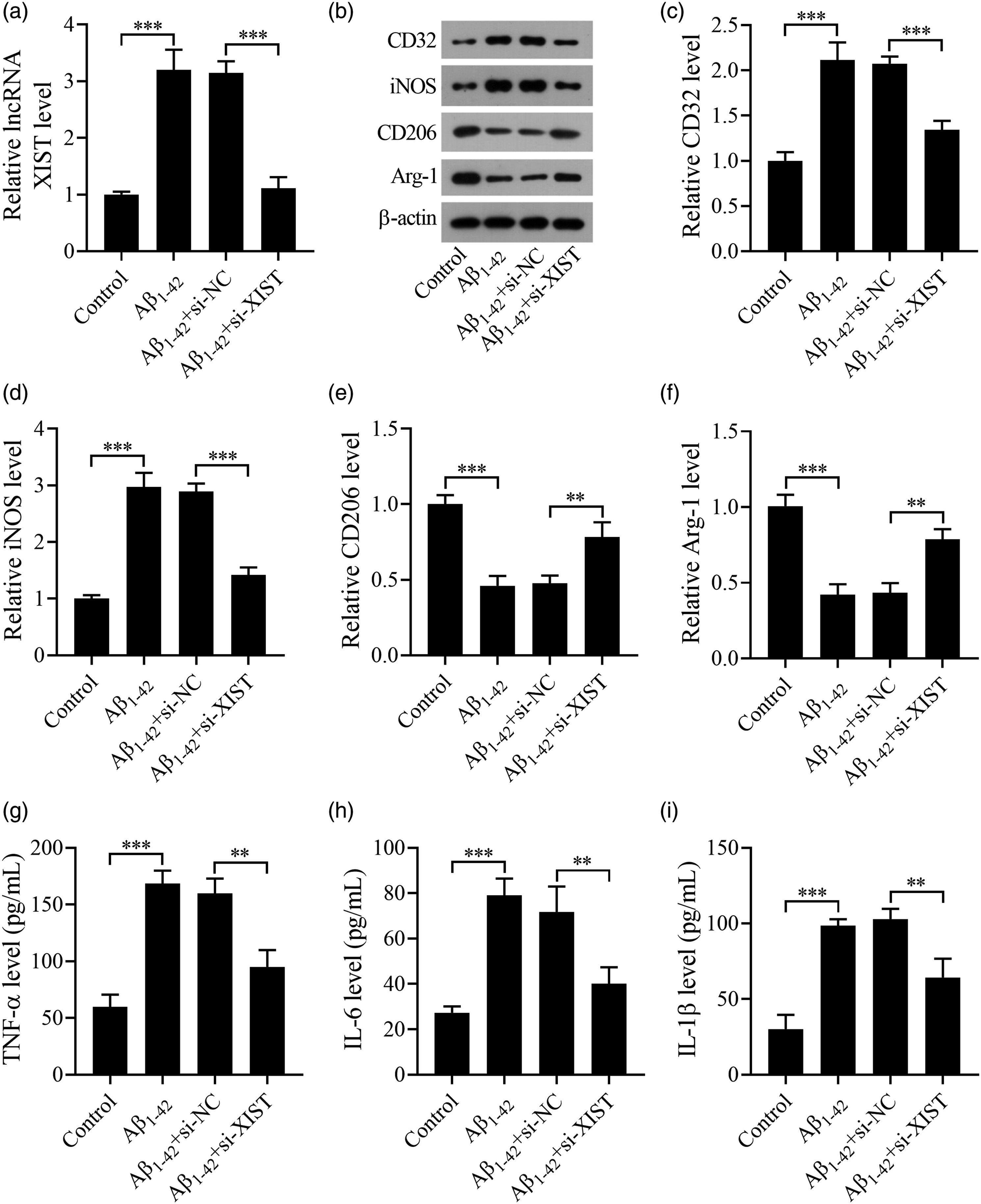
XIST knockdown attenuates Aβ1–42-induced microglia-mediated neurotoxicity
To mimic the Aβ1–42-induced microglia-mediated neurotoxicity in AD, the BV2 cells were transfected with si-NC or si-XIST and then stimulated with Aβ1–42 (10 μM) for 24 h, and afterward the mediums were collected as conditioned mediums. The conditioned mediums were used to incubate HT22 cells for 48 h. Results showed that the percent of TUNEL-positive cells (Figures 4(a) and (b)) and the caspase-3 activity (Figure 4(c)) were increased in HT22 cells in the Aβ1-42 group compared with the control group, while this influence was partially eliminated by downregulation of XIST. As displayed in Figure 4(d), the viability of HT22 cells was reduced in the Aβ1-42+sh-NC group, but improved in the Aβ1-42+sh-XIST group. There was no statistical difference between the Aβ1-42 group and the Aβ1-42+si-NC group. In a word, knockdown of XIST diminished Aβ1–42-induced microglia-mediated neurotoxicity. XIST knockdown attenuates Aβ1-42-induced microglia-mediated neurotoxicity. (a, b) The apoptosis of HT22 cells was assessed by TUNEL assay. Scale bar: 50 μM. (c) The caspase-3 activity of HT22 cells was detected by the caspase-3 activity kit. (d) The viability of HT22 cells was measured by CCK8 assay. **p < .01 and ***p < .001. All experiments were independently repeated three times.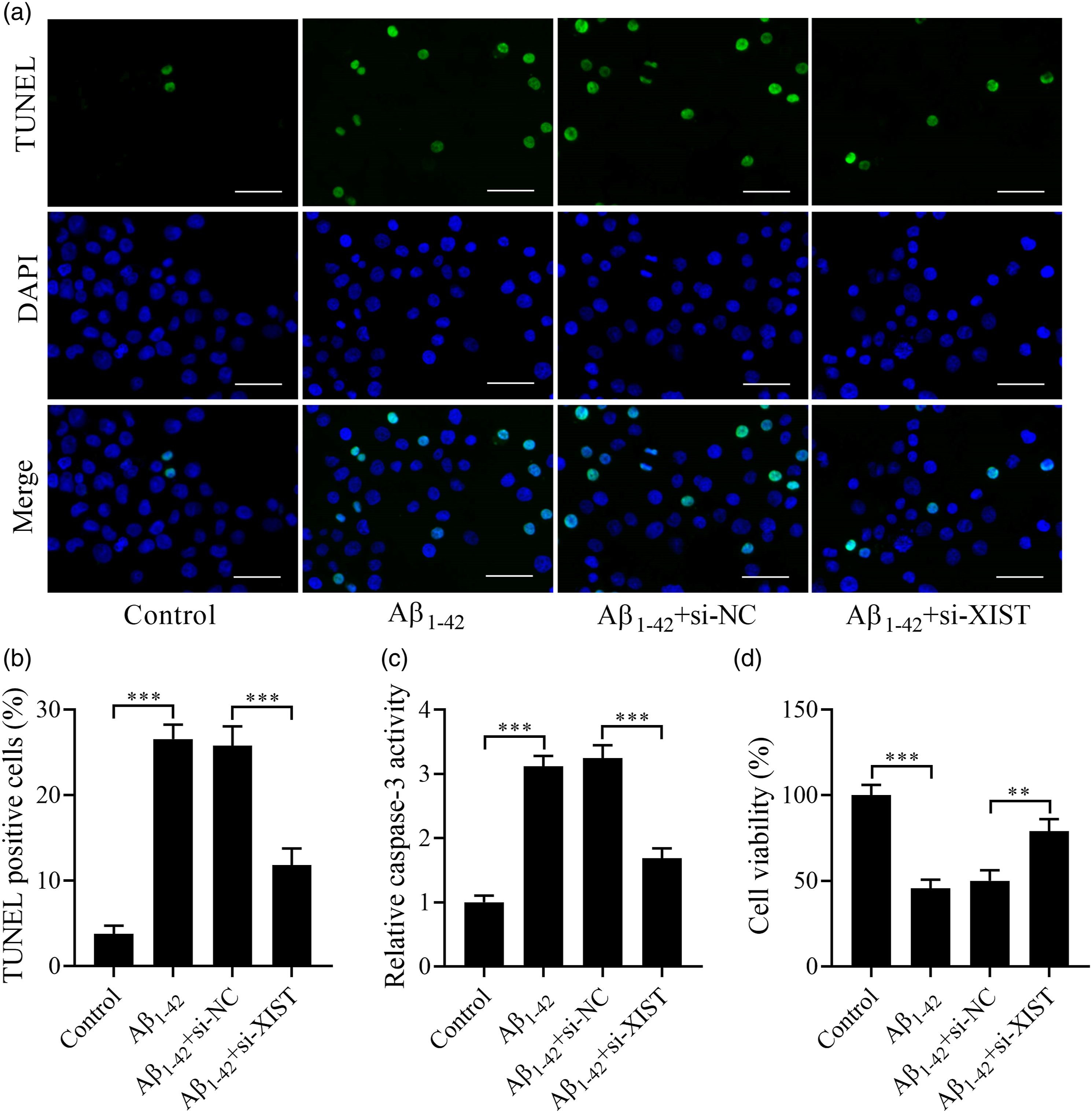
miR-107 and the PI3K/Akt signaling are involved in the effect of XIST on Aβ1–42-induced microglia-mediated neurotoxicity
In this part, we predicted the downstream miRNA targets of XIST. The 186 and 454 miRNA targets were, respectively, predicted via miRcode and starbase. In addition, AD 24 related miRNA targets (score ≥10) were predicted by GeneCards. Only one overlapping target, miR-107, was found by Venn diagram analysis (Figure 5(a)). Then, we explored the downstream gene targets of miR-107. The 1252 and 4288 gene targets were correspondingly predicted by MicroT-CDS and starbase. Moreover, AD 424 related gene targets (score ≥10) were predicted by GeneCards. There were 33 overlapping gene targets (Figure 5(b)). Next, KEGG analysis was carried out on the 33 targets and the pathways (p < .01) were displayed in Figure 5(c). The targets were associated with the PI3K-Akt signaling pathway (Figure 5(c)). Herein, BV2 cells were grouped into the control, Aβ1-42, Aβ1-42+si-NC, Aβ1-42+si-XIST, Aβ1-42+si-XIST + anti-NC, Aβ1-42+si-XIST + anti-miR-107, Aβ1-42+si-XIST + LY294002 groups. The treatment methods of the control, Aβ1-42, Aβ1-42+si-NC, and Aβ1-42+si-XIST groups were consistent with those in 3.3. The BV2 cells in the Aβ1-42+si-XIST + anti-NC or Aβ1-42+si-XIST + anti-miR-107 group were co-transfected with corresponding gene molecules, and then stimulated with Aβ1-42 (10 μM) for 24 h. The BV2 cells in the Aβ1-42+si-XIST + LY294002 group were pretreated with LY294002 (10 μM) for 1 h and then subjected to si-XIST transfection. After transfection, BV2 cells were stimulated with Aβ1-42 (10 μM) for 24 h. We found that the rates of p-P13 K/P13 K (Figures 5(d) and (e)) and p-Akt/Akt (Figures 5(d) and (f)) in BV2 cells were reduced by Aβ1-42 treatment, while this effect was attenuated by XIST knockdown. In addition, both inhibition of miR-107 and the PI3K/Akt signaling partially reversed XIST silencing-mediated effect on the rates of p-P13 K/P13 K and p-Akt/Akt in Aβ1-42 treated BV2 cells. Therefore, we concluded that XIST could regulate miR-107 and the PI3K/Akt signaling in Aβ1-42 treated BV2 cells. XIST regulates miR-107 and the PI3K/Akt signaling in Aβ1-42-stimulated microglial cells. (a) Venn diagram of overlapped miRNA targets in XIST and AD. (b) Venn diagram of overlapped gene targets in miR-107 and AD. (c) KEGG analysis of 33 gene targets. (d) The protein levels of p-P13 K, P13 K, p-Akt, and Akt were detected by western blot. (e) The rate of p-P13 K/P13 K. (f) The rate of p-Akt/Akt. **p < .01 and ***p < .001. All experiments were independently repeated three times.
Inhibition of miR-107 or the PI3K/Akt signaling attenuates XIST silencing-mediated regulation of microglial M1/M2 polarization in Aβ1-42-stimulated BV2 cells
The miR-107 level in Aβ1-42-stimulated BV-2 cells was increaed by si-XIST transfection and reduced by anti-miR-107 co-treatment. Compared with the si-XIST + anti-miR-107 group, the miR-107 level was increased in the si-XIST + LY294002 group (Figure 6(a)). The levels of CD32 and iNOS (Figures 6(b)–(d)) were decreased and the Arg-1 and CD206 levels (Figure 6(b), (e), and (f)) were increased in Aβ1-42-stimulated BV-2 cells after XIST knockdown, but these changes were weakened by downregulation of miR-107 or inhibition of the PI3K/Akt signaling. Moreover, the TNF-α (Figure 6(g)), IL-6 (Figure 6(h)), and IL-1β (Figure 6(i)) levels in Aβ1-42-stimulated BV-2 cells were reduced by si-XIST transfection, and elevated by anti-miR-107 or LY294002 co-treatment. Thus, these results indicated that inhibition of miR-107 or the PI3K/Akt signaling attenuated the effect of XIST silencing on Aβ1-42-caused imbalance of microglial M1/M2 polarization and secretion of proinflammatory factor levels. Inhibition of miR-107 or the PI3K/Akt signaling reverses the effect of XIST silencing on microglial M1/M2 polarization in Aβ1-42-stimulated BV2 cells. (a) The level of miR-107 in BV2 cells was determined by qRT-PCR. (b–f) The protein levels of CD32, iNOS, Arg-1, and CD206 were detected by western blot. (g–i) The levels of TNF-α, IL-6, and IL-1β were measured by ELISA. *p < .05, **p < .01, and ***p < .001. All experiments were independently repeated three times.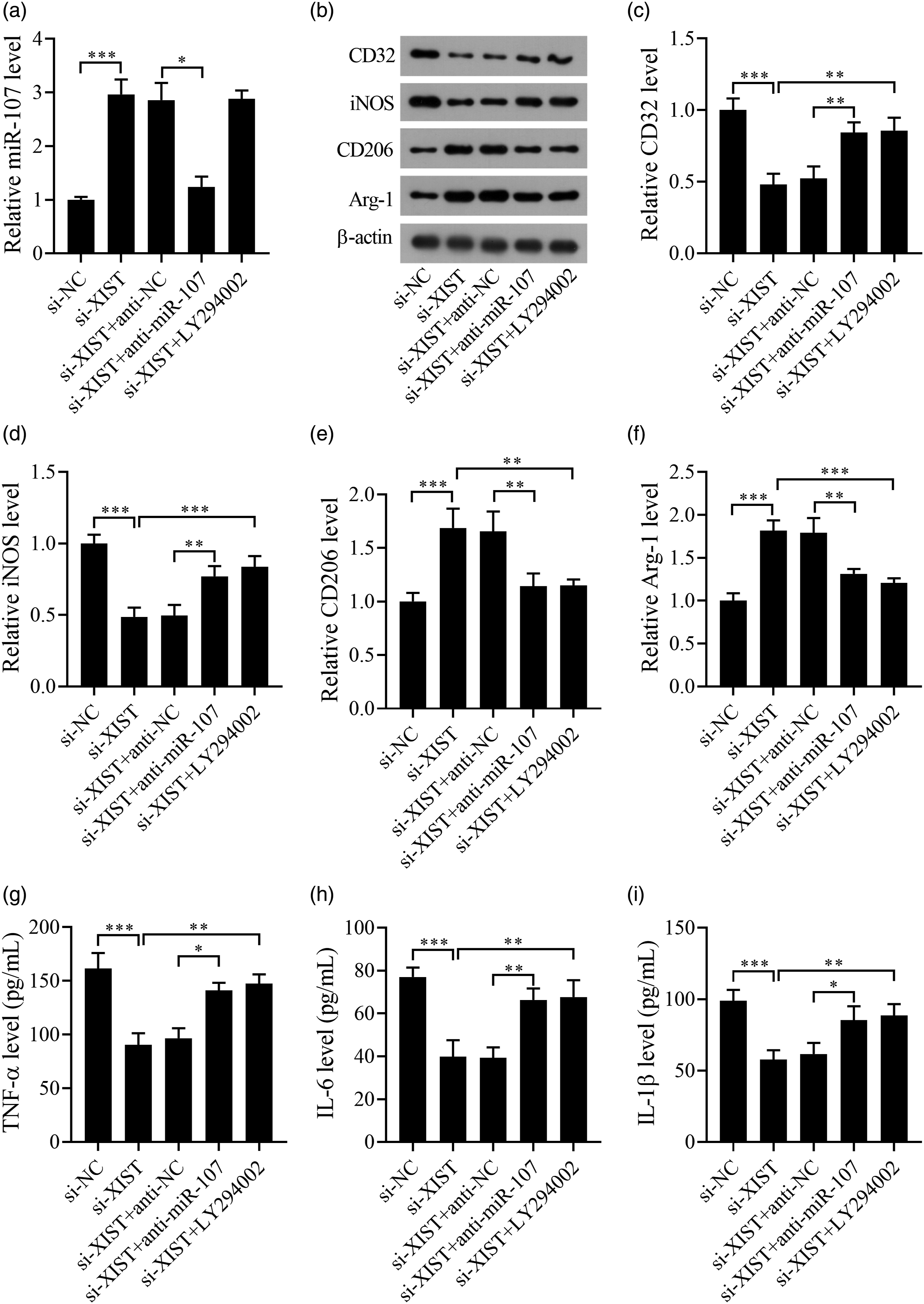
Inhibition of miR-107 and the PI3K/Akt signaling attenuates the protection of XIST silencing on Aβ1-42-induced microglia-mediated neurotoxicity
In this part, the mediums of Aβ1-42-stimulated BV2 cells were collected as conditioned mediums. The conditioned mediums were used to incubate HT22 cells for 48 h. Results showed that the apoptosis of HT22 cells (Figures 7(a)–(c)) were reduced and cell viability (Figure 7(d)) was elevated by si-XIST transfection, but these changes were attenuated by anti-miR-107 or LY294002 co-treatment. The molecular mechanism underlying XIST regulating AD development was shown in Figure 8. These findings suggested that inhibition of miR-107 or the PI3K/Akt signaling could attenuate the effect of XIST silencing on Aβ1-42-induced microglia-mediated neurotoxicity. Inhibition of miR-107 or the PI3K/Akt signaling reverses the effect of XIST silencing on Aβ1-42-induced microglia-mediated neurotoxicity. (a, b) The apoptosis of HT22 cells was assessed by TUNEL assay. Scale bar: 50 μM. (c) The caspase-3 activity of HT22 cells was detected by the caspase-3 activity kit. (d) The viability of HT22 cells was measured by CCK8 assay. *p < .05, **p < .01 and ***p < .001. All experiments were independently repeated three times. A schematic diagram illustrates the regulatory mechanism of XIST-mediated neuroinflammation and neurotoxicity.

Discussion
Our study has discovered a new role of XIST in the development of AD. We observed an increase in XIST content in the hippocampus of APP/PS1 mice. Downregulation of XIST in the hippocampus enhanced the spatial learning and memory capability of APP/PS1 mice. Besides, knockdown of XIST reduced the abundance of Aβ1-42 and reduced cell apoptosis in the hippocampus of APP/PS1 mice. Moreover, XIST silencing abridged activation of microglia and microglial M1 polarization, but contributed to microglial M2 polarization in APP/PS1 mice. In Aβ1-42-treated BV-2 cells, XIST knockdown suppressed microglial M1 polarization and proinflammatory factor levels, while facilitated microglial M2 polarization. XIST knockdown attenuated Aβ1-42-induced microglia-mediated cell apoptosis and enhanced cell viability in HT22 cells. The predicted results of bioinformatics analysis showed that miR-107 is possibly a target of XIST, and the targets of miR-107 and AD were enriched in the PI3K/Akt signaling pathway. XIST could down-regulate miR-107 and inhibit the PI3K/Akt signaling in Aβ1-42-stimulated BV2 cells. We found that inhibition of miR-107 or the PI3K/Akt signaling reversed the effect of XIST silencing on microglial M1/M2 polarization, proinflammatory factor levels, and Aβ1-42-induced microglia-mediated neurotoxicity. Consequently, our consequences suggest that the XIST/miR-107/PI3K/Akt signaling pathway play a crucial role in the advancement of AD, indicating that silencing of lncRNA XIST and/or miR-107 upregulation might be a potential therapy for the management of AD.
LncRNAs play a key role in microglia and neurocyte, which modulates the development of neurodegenerative disorders, including AD.33–35 For instance, Gu et al. 36 demonstrated that the level of XIST was heightened in the rat spinal cord damage model and silencing of XIST exerted a protective effect against spinal cord injury. Additionally, our previous research revealed that the downregulation of XIST can attenuate Aβ25-35-induced toxicity and apoptosis in rat hippocampal neurons by modulating miR-132. 16 Yan et al. 37 also confirmed that the level of XIST was elevated in the mouse and cell models of AD. Downregulation of XIST has been found to reduce Aβ-induced neuronal inflammation and injury by epigenetically inhibiting the Aβ-degrading enzyme NEP. In our research, we observed that the level of XIST was increased in the hippocampus of APP/PS1 mice and Aβ-treated BV-2 cells, which was consistent with the results of Yan et al. 37 As the innovation of our study, we demonstrated for the first time that regulation of microglial M1/M2 polarization was an important mechanism underlying that lncRNA XIST participated in the development and progression of AD. Our findings contribute to further understanding of the pathogenesis of AD.
The hippocampus is a vital structure in the brain and plays a crucial role in spatial learning and memory. In AD patients, the hippocampus is the most affected part of the brain. Aβ is the main component of amyloid cores in senile plaques, and the accumulation and deposition of Aβ is a significant cause of AD. 38 Herein, our study found that the downregulation of XIST in the hippocampus improved the spatial learning and memory capability of APP/PS1 mice. Additionally, we confirmed that the cell morphology in the hippocampal tissue structure of APP/PS1 mice was altered, and the level of Aβ1-42 was increased. Our findings showed that downregulation of XIST attenuated these changes. These results suggest that knockdown of XIST can constrain the progression of AD in APP/PS1 mice.
The study conducted by Malvindar et al. revealed that the content of Iba1 in AD was increased. 39 In this study, the Iba1 level was increased in APP/PS1 mice, which was consistent with the findings of Malvindar et al. 39 Additionally, we firstly demonstrated that XIST silencing reduced Iba1 level and microglial M1 polarization, but facilitated microglial M2 polarization in APP/PS1 mice. Furthermore, we treated the BV2 cells with Aβ1-42 to mimic the AD cell environment. It was shown that XIST knockdown repressed microglial M1 polarization and proinflammatory factor levels, but facilitated microglial M2 polarization in vitro. Moreover, XIST knockdown attenuated Aβ1-42-induced microglia-mediated neurotoxicity. Similarly, Chen et al. demonstrated that the content of miR-107 was reduced in the brain tissue of AD patients. The modulation of the PI3K/Akt pathway by MiR-107 lessened injury in an AD cell model. 40 Finally, we firstly confirmed that XIST could suppress miR-107 and the PI3K/Akt signaling in Aβ1-42 treated BV2 cells. Inhibition of miR-107 or the PI3K/Akt signaling reversed the effect of XIST silencing on microglial M1/M2 polarization, proinflammatory factor levels, and Aβ1-42-induced microglia-mediated neurotoxicity. These results were consistent with the findings of Chen et al. 40
Limitations of this study
There are still some limitations in this paper. For animal experiments, we did not conduct power analysis to estimate the sample size of mice. Therefore, more effort should be done to improve animal welfare and minimize the number of animals used. We detected the expression level of lncRNA XIST in both animal and cellular AD models, but its abnormal expression was not validated in clinical samples of AD patients. Further experiments should be performed to verify whether miR-107 acts a direct target gene of lncRNA XIST. Our next research focus is to reveal the upstream regulation mechanism of lncRNA XIST/miR-107 axis in the progression of AD.
Conclusion
In summary, our findings indicate that XIST was upregulated in the hippocampus of APP/PS1 mice and Aβ-treated BV-2 cells. Downregulation of XIST reduced Aβ1-42-induced microglia-mediated neurotoxicity by regulating microglial M1/M2 polarization. Furthermore, the miR-107/PI3K/Akt pathway axis was involved in the function of XIST in AD progression. Our findings contribute to further understanding of the mechanism of microglia-mediated neuroinflammation in neuronal microenvironment during the progression of AD. This research identifies a potential therapeutic approach for AD management.
Footnotes
Author contributions
We declare that this work was done by the authors named in this article and all liabilities pertaining to claims relating to the content of this article will be borne by the authors.
Kunpeng Zhao conceived and designed the study.
Xinyu Wang and Meiqi Shao collected the data. Chenyang He and Fuqiang Yuan analyzed the data. Kunpeng Zhao wrote the manuscript. All authors read and approved the manuscript for publication.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the This paper was funded by the Henan Provincial Science and Technology Research Program (NO. 212102310588).
Ethics statement
This study was approved by an institutional review board of The Second Affiliated Hospital of Xinxiang Medical University, Henan Mental Hospital. Ethical approval for this study was obtained from “The Ethics Committee of Henan Mental Hospital (APPROVAL NUMBER/2020-1011)”.
Animal welfare
The present study followed the Guide for the Care and Use Laboratory Animals of Henan Mental Hospital and complied with Regulations of the People’s Republic of China for the Administration of laboratory animals.