
Abstract
Objectives
We investigated the interactions among DHX9, phosphorylated DHX9, R-loops, and DNA damage to clarify the mechanism by which phosphorylated DHX9 inhibited lung adenocarcinoma progression.
Methods
Used PC-9 and 2BS cells divided into control, siDHX9, OE-DHX9, siDHX9 + OE-RNase H1, OE-PKA, DHX9-S279A, 6-22 Amide, and DHX9-S279A + OE-RNase H1 groups. Assays included quantitative real-time polymerase chain reaction (qRT-PCR), WB (DHX9, γH2AX, Rad51, pCtIP), EdU/CCK-8 (proliferation), TUNEL/flow cytometry (apoptosis), comet assay (DNA damage), CldU/IdU (replication), DRIP-qPCR (R-loops). Nude mice xenografts (control, siDHX9, DHX9-S279E, DHX9-S279A) assessed tumor growth, Ki67, R-loops, DNA damage, and replication.
Results
DHX9 was highly expressed in multiple cancer tissues and lung cancer cell lines, with higher messenger RNA levels in PC-9 than in 2BS cells. Compared with PC-9, siDHX9 reduced proliferation and increased apoptosis, while OE-DHX9 exerted opposite effects. siDHX9 increased DNA damage (with corresponding changes in γH2AX, Rad51, and pCtIP levels), reduced replication (rescued by OE-RNase H1), and elevated R-loops; OE-DHX9 showed opposite effects on damage and R-loops. OE-PKA increased R-loops and damage, and reduced replication, while DHX9-S279A or 6-22 Amide decreased these and 6-22 Amide also increased replication versus PC-9/OE-PKA. DHX9-S279A increased proliferation, with DHX9-S279A + OE-RNase H1 further enhancing this and reducing apoptosis. In vivo, siDHX9 and DHX9-S279E reduced tumor volume/mass and Ki67, increased R-loops, damage, and γH2AX/Rad51/pCtIP, and inhibited replication; DHX9-S279A showed opposite effects versus these groups, with no significant tumor difference versus PC-9 and higher replication versus both.
Conclusions
Phosphorylated DHX9 might enhance DNA damage by suppressing R-loop resolution, ultimately inhibiting the proliferation of lung adenocarcinoma cells.
Introduction
Despite ongoing advancements in targeted therapy and immunotherapy for non-small cell cancer (NSCLC)—which have improved patients’ progression-free survival and objective response rates—the 5-year survival rate remains below 20%. 1 DNA damage is tightly linked to tumor biological functions: it acts as a key trigger for tumor suppression by activating the DNA damage response pathway, thereby effectively inhibiting cancer cell proliferation. 2 In NSCLC, AXL inhibition-induced DNA damage and replication stress enhance sensitivity to ATR inhibitors. 3 >DHX9, a versatile DExH-box RNA helicase, plays a pivotal role in maintaining genomic stability and regulating tumorigenesis.4,5 >It is frequently overexpressed in various malignancies, including lung adenocarcinoma, where it acts as a pro-oncogenic factor by modulating transcriptional activation (e.g. epidermal growth factor receptor (>EGFR) pathway) and promoting epithelial-mesenchymal transition (EMT).6–8 Mechanistically, DHX9 is critical for resolving R-loops—three-stranded nucleic acid structures consisting of an RNA:DNA hybrid and a displaced single-stranded DNA.6,9 By efficiently unwinding R-loops, DHX9 prevents replication fork stalling and subsequent DNA double-strand breaks. 10 Furthermore, the helicase activity and localization of DHX9 are tightly regulated by post-translational modifications, particularly phosphorylation, which governs its interaction with DNA repair factors and its ability to safeguard the genome against stress. 9 R-loops can impede gene transcription and induce DNA double-strand breaks, compromising genomic stability.11,12 DHX9 unwinds R-loops approximately five to seven times more efficiently than equivalent DNA and RNA forks. 13 However, DHX9 activity is regulated by phosphorylation; ATR-mediated phosphorylation at S321 represents a critical mechanism governing its activity in DNA damage responses and R-loop resolution. 9 Notably, the effects of phosphorylation at different sites on R-loop homeostasis and lung cancer progression remain undefined. Additionally, whether DHX9 and phosphorylated DHX9 mediate DNA damage via R-loop regulation to ultimately impact lung adenocarcinoma cell proliferation remains unclear. We hypothesize that in lung cancer, DHX9 phosphorylation impairs its ability to resolve R-loops, leading to increased R-loop-mediated genomic instability and cell cycle arrest, which ultimately suppress tumor growth and metastasis.
Materials and methods
Cell lines and cell culture
Human embryonic lung diploid 2BS cells were kindly provided by the Institute of Basic Medical Sciences, Peking Union Medical College; PC-9 human lung adenocarcinoma cells were purchased from Shanghai Fuheng Biotechnology Co., Ltd. The 2BS cells were cultured in EMEM and the PC-9 cells in Roswell Park Memorial Institute (RPMI)-1640, both supplemented with 10% fetal bovine serum (FBS) (Zhejiang Tianhang Bio-Technology Co., Ltd). Experiments were performed using cells harvested at the logarithmic growth phase.
Small interfering RNAs, plasmids, and cell transfections
DHX9-targeting small interfering RNA (siRNA) (siDHX9) and negative control siRNA (siNC) were designed and synthesized by Ribobio (Guangzhou, China). Overexpression vectors and control plasmid (pcDNA3.1) were constructed by GenePharma (Shanghai, China). Cells were transfected with siRNAs or plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer's protocol, then divided into groups: control(PC-9), siDHX9, OE-DHX9, siDHX9 + OE-RNase H1, OE-PKA(DHX9-interacting phosphokinase), DHX9-S279A(inhibiting phosphorylation), 6-22 Amide (phosphokinase inhibitor), DHX9-S279A + OE-RNase H1, and DHX9-S279E (activating phosphorylation). Cells were processed for subsequent experiments 24 h post-transfection. The phosphokinase inhibitor 6-22 amide (CAS#121932-06-7) was added to the 6-22 Amide group 12 h before experimentation.
Quantitative real-time polymerase chain reaction
Total RNA was isolated from cells using TRIzol reagent (Invitrogen) per the manufacturer's protocol. Equal amounts (1 μg) of total RNA were reverse-transcribed to cDNA with the PrimeScript RT Master Mix Kit (Takara-Bio, Dalian, China). Real-time polymerase chain reaction (PCR) was performed on an ABI 7900 system (Applied Biosystems, Foster City, CA), with relative quantification via the 2-ΔΔCt method. GAPDH served as the internal control for DHX9 messenger RNA (mRNA) expression.
EdU staining assay
The EdU Cell Proliferation Kit (Promega) was used per the supplier's protocol. In a 96-well plate, 5 × 103 logarithmic-phase cells were seeded and cultured to normal growth. Once 80% confluent, the cells were treated with 10 μM EdU for 24 h, fixed with 4% paraformaldehyde, and stained sequentially with Apollo 567 and Hoechst 33342 following kit instructions. Blue and red cells in three random fields were counted under a fluorescence microscope.
Cell Counting Kit-8 assay
A 100 μL cell suspension (pretreated with PBS and 0, 0.2, 0.4, or 0.8 μg/mL NETs) was seeded in 96-well plates at 5000 cells/well (five plates/group). Plates were cultured at 37°C with 5% CO₂ for 12, 24, 48, or 72 h. At each time point, 10 μL Cell Counting Kit-8 (CCK-8) solution was added, and plates were incubated for 4 h. Absorbance at 450 nm was measured using a microplate reader, with data collected and statistically analyzed.
TUNEL staining assay
Logarithmic-phase-phase cells were seeded on coverslips in 12-well plates. After intervention, cells were fixed, permeabilized, and blocked, then subjected to TUNEL staining using the In Situ Cell Death Detection Kit (Thermofisher). Freshly prepared 50 μL TUNEL reaction solution (1:9 ratio, ice-handled) was added dropwise, followed by moist, light-protected incubation at 37°C for 60 min. After phosphate-buffered saline (PBS) washing, DAPI was added for nuclear counterstaining. Following rinsing, an anti-fluorescence quenching agent was applied to slides, coverslips were inverted onto them for mounting, and staining was observed under a microscope at 400× magnification.
Flow cytometry assay
Cells were seeded in 6-well plates at 1.0 × 105 cells/mL and incubated overnight. When density reached ∼(1-5) × 106 cells/mL, cells were harvested, centrifuged at 1000 r/min for 5 min, and the supernatant was discarded. Cell pellets were washed with 3 mL PBS, centrifuged, and the PBS was removed; ice-cold 70% ethanol was added for fixation at 4°C for 2 h. After fixation, cells were centrifuged to remove fixative, resuspended in 3 mL PBS for 5 min, and filtered through a 400-mesh sieve. Filtered cells were centrifuged (1000 r/min, 5 min) to remove PBS, resuspended in an annexin binding buffer containing Annexin V and PI, and incubated at room temperature for 15 min in the dark. Apoptosis was analyzed using a BD LSRFortessa Cell Analyzer (BD Biosciences).
Comet assay
Logarithmic-phase cells were harvested, trypsin-digested, and resuspended in 1640 medium with 10% FBS to prepare a cell suspension, which was seeded into 6-well plates. After 24 h of adherence, cells were collected and adjusted to 5 × 105 cells/mL. A 400 μL aliquot of 7.5% normal-melting-point agarose gel was spread evenly on glass slides and solidified at 4°C for 20 min. Then 30 μL cell suspension (5 × 105 cells/mL) was mixed thoroughly with 70 μL 7.5% low-melting-point agarose gel, and the mixture was spread over the first-layer gel, followed by incubation at 4°C for 20 min. Samples were lysed in an alkaline lysis buffer at 4°C for 2.5 h, rinsed with PBS, and incubated in electrophoresis buffer at 4°C for 20 min for DNA unwinding. Electrophoresis was performed at 20 V and 200 mA for 20 min. After PBS rinsing, samples were neutralized in a buffer solution at 4°C for 20 min, stained with 2 μg/mL ethidium bromide, rinsed with double-distilled water, and visualized under a fluorescence microscope. Over 100 random comet images were captured and analyzed using CASP software (http://casplab.com/).
Western blotting analysis
Protein lysates were prepared by lysing cells in radio-immunoprecipitation assay buffer with protease inhibitors (kept on ice to preserve integrity). Lysates were centrifuged at 1400 g for 15 min at 4°C to extract proteins, whose concentration was quantified via BCA assay. Proteins were separated by SDS-PAGE, transferred to PVDF membranes, and blocked in 5% skim milk. Membranes were incubated overnight at 4°C with primary antibodies against γH2AX(ab26350), Rad51(ab109107), pCtIP(ab284851), and β-actin(ab213262), followed by 1 h room-temperature incubation with secondary antibodies. Chemiluminescent signals were detected using ChemiDoc XRS (Bio-Rad), X-ray films developed automatically, and bands analyzed via Quantity One software.
CldU/IdU DNA fiber pulse labeling assay
Cell culture medium containing FBS and RPMI was used to culture cells in sterile flasks at 37°C with 5% CO2. Cells(5 × 105/well) were seeded in 6-well plates for slide preparation. CldU and IdU reagents were mixed at 60°C until nucleotide analog UES dissolved, then incubated with cells for 20 min before placing on ice. Fiber lysis solution (200 mM Tris-HCl, 50 mM EDTA pH 7.5, 0.5% SDS) was prepared; 7 μL was gently mixed with cell lysate and incubated for 2 min. Slides were tilted at 15 degrees to allow fiber spreading, then dried, and the fibers were stretched until staining lines disappeared, then marked with pencil for later localization. DNA fiber bundles were measured and replication structures counted, selecting only clearly visible fibers not extending to image edges and suitable for analysis.
DRIP-qPCR assay
Confluent cells were washed twice with 1×PBS, scraped with 3 mL 1×PBS, pipetted to form a suspension, and centrifuged at 1200 rpm for 5 min. The pellet was resuspended in 1.2 mL DNA lysis buffer, aliquoted into 1.5 mL tubes (300 μL each) with 10 μL proteinase K, and incubated at 56°C for 5 h. Equal volumes of Tris-saturated phenol were added, mixed by inversion, shaken at room temperature for 5 min, and centrifuged at 12,000 rpm at 4°C for 10 min; the upper aqueous phase was transferred. Equal volumes of phenol-chloroform (25:24:1) were added, mixed, shaken, and centrifuged as above; the upper phase was transferred, 2.5 volumes of pre-chilled(−20°C) ethanol were added, and centrifuged at 12,000 rpm at 4°C for 7 min. Supernatant was removed, 400 μL pre-chilled 75% ethanol was added then removed, and the DNA pellet was air-dried in a clean bench and dissolved in TE. For 50 μg extracted DNA, 20 U EcoRI, 20 U HindIII, 20 U XbaI, 25 U SspI, 10 U BsrGI, and buffer 2.1 were added for digestion at 37°C for 5 h (control group: +5 U RNase H). Digested DNA was purified as above. 10 μg recovered DNA was diluted to 900 μL with TE, 100 μL 10×DRIP buffer added. 20 μL protein G Dynabeads were pre-blocked at 4°C for 40 min, then 10 μg S9.6 antibody added for overnight blocking at 4°C. After centrifugation at 1500 rpm at 4°C for 3 min, the pellet was resuspended in 1×DRIP buffer, centrifuged again (repeated), then resuspended in 1×DRIP buffer+330 mM NaCl and centrifuged. Supernatant was removed, DRIP elution buffer added, incubated at 65°C for 45 min, and the supernatant used for qPCR or stored at −20°C.
Tumor xenograft model in nude mice
Male BALB/c nude mice (6 weeks old) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. The mice were randomly divided into four groups (n = 6 per group): control, siDHX9, DHX9-S279E, and DHX9-S279A. Stably transfected PC-9 cells (5 × 107 cells), which were engineered to stably express siDHX9, DHX9-S279E, or DHX9-S279A using lentiviral vectors, were injected subcutaneously into the posterior flank. Food intake and body weight were monitored; tumor volume was measured weekly for 4 weeks using the formula: length × width2/2. At sacrifice, tumors were excised, weighed, and stored at −80°C for further analysis. All animal experiments were approved by the Animal Ethics Committee of The First Affiliated Hospital of Xi’an Medical University (Approval No. XYYFYLL-KTSB-2023-10) and were conducted in accordance with the institutional guidelines for the care and use of laboratory animals.
Immunohistochemistry assay
Paraffin sections were dewaxed and hydrated via immersion in xylene, then 100%, 95%, 90%, 80%, and 70% ethanol. After rinsing in PBS (pH 7.4) three times (3 min each), sections were soaked in citric acid solution, heated to 95°C on an induction cooker for 20 min for antigen retrieval, and rinsed again in PBS (3 × 3 min). They were incubated with peroxidase inhibitor at room temperature for 10 min, rinsed in PBS (3 × 3 min), and blocked with serum. After removing serum, primary antibody (rabbit anti-human Ki67 monoclonal antibody, 1:100, Abcam) was added dropwise, and sections were incubated overnight at 4°C in a wet chamber. Following PBS rinses (3 × 5 min), goat anti-rabbit secondary antibody (1:100, Abcam) was added dropwise, then sections were rinsed in PBS (3 × 3 min). Horseradish peroxidase was added dropwise, followed by PBS rinses (3 × 3 min). DAB was added dropwise, and sections were washed with distilled water, then dehydrated, cleared, and mounted.
Statistical analysis
Data analysis was performed using SPSS 20.0, with results expressed as mean ± standard deviation (x ± s). Comparisons between two groups were analyzed by t-test, and among multiple groups by multivariate analysis of variance. A P-value <0.05 indicated statistical significance.
Results
DHX9 was enriched in cancer cell lines
We accessed the Protein Atlas database (https://www.proteinatlas.org) to query DHX9 expression in various lung cancer cell lines, with RNA expression data presented as normalized transcript per million (nTPM) values. Among 232 lung cancer cell lines, PC9 cells showed a DHX9 RNA nTPM of 115 (Figure S1(a), only available online). Quantitative real-time polymerase chain reaction (qRT-PCR) detection revealed significantly higher DHX9 mRNA expression in human lung adenocarcinoma PC-9 cells than in human embryonic lung diploid 2BS cells (Figure S1(b), only available online).
Functional roles of DHX9 in lung adenocarcinoma cells: promoting proliferation and suppressing apoptosis
To investigate whether DHX9 promotes proliferation and inhibits apoptosis in PC-9 cells, we downregulated DHX9 via siRNA transfection and overexpressed it using pcDNA3.1-DHX9. The transfection efficiency was verified by qRT-PCR and Western blotting, which confirmed significant downregulation of DHX9 mRNA and protein levels in the siDHX9 group and significant upregulation in the OE-DHX9 group compared to their respective controls (Figure S2(a) and (b)), only available online). We then assessed effects via EdU staining, CCK-8 assay, TUNEL staining, and flow cytometry. EdU staining showed significantly lower proliferative activity in the siDHX9 group versus the PC-9 group, with higher activity in the OE-DHX9 group versus both the siDHX9 and PC-9 groups (Figure 1(a) and (b)). CCK-8 assay revealed significantly lower optical density (OD) values in the siDHX9 group versus the PC-9 group, and higher values in the OE-DHX9 group versus both controls (Figure 1(c)). TUNEL staining and flow cytometry both demonstrated significantly higher apoptosis rates in the siDHX9 group versus the PC-9 group, with lower rates in the OE-DHX9 group versus both the siDHX9 and PC-9 groups (Figure 1(d) to (g)).
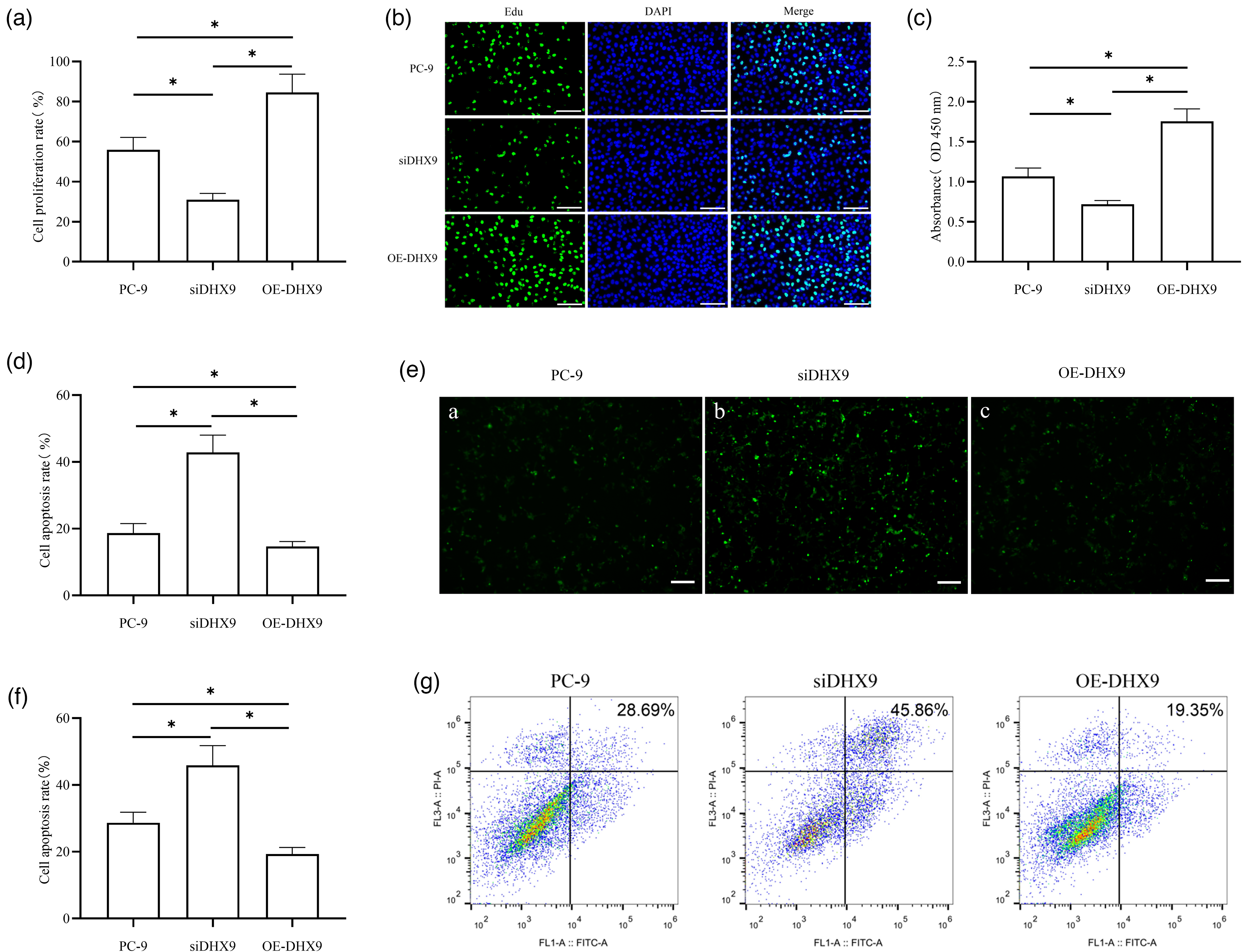
Functional roles of DHX9 in lung adenocarcinoma cells. The effect of DHX9 on lung adenocarcinoma cell proliferation was detected by EdU staining (a) and (b) and CCK-8 assay (c). The cell proliferation was detected by CCK-8 assay. The absorbance (OD) values presented represent the data collected at 48 h post transfection. The effect of DHX9 on lung adenocarcinoma cell apoptosis was detected by TUNEL staining (d) and (e) and flow cytometry (f) and (g). Apoptosis was assessed by TUNEL staining and flow cytometry at 48 h post-transfection. *P < 0.05.
Mechanism of DHX9 in lung adenocarcinoma cells: promoting R-loop resolution and reducing DNA damage
To investigate whether DHX9 promotes R-loop resolution and reduces DNA damage in lung adenocarcinoma cells, cells were transfected and divided into control, siDHX9, OE-DHX9, and siDHX9 + OE-RNase H1 groups. DNA damage was assessed by comet assay, with Western blotting detecting the DNA damage marker γH2AX and the homologous recombination repair proteins Rad51 and pCtIP; DNA replication status was measured via CldU/IdU fiber labeling, and R-loops accumulation via DRIP-qPCR. Comet assay showed a significantly longer tail length and a higher comet rate in the siDHX9 group versus the PC-9 group, with opposite effects in the OE-DHX9 group versus both the PC-9 and siDHX9 groups (Figure 2(a) to (c)). Western blotting revealed significantly higher γH2AX, Rad51, and pCtIP levels in the siDHX9 group versus the PC-9 group, and lower levels in the OE-DHX9 group versus both controls (Figure 2(d) to (g)). CldU/IdU labeling indicated significantly shorter DNA fibers in the siDHX9 group versus the PC-9 group (no significant difference between the OE-DHX9 and PC-9 groups), with longer fibers in the siDHX9 + OE-RNase H1 group versus the siDHX9 group, suggesting improved replication after RNase H1 overexpression (Figure 2(h) and (i)). DRIP-qPCR showed significantly higher DRIP enrichment in the siDHX9 group versus the PC-9 group, lower enrichment in the OE-DHX9 group versus both controls, and reduced enrichment in the siDHX9 + OE-RNase H1 group versus the siDHX9 group, indicating that RNase H1 overexpression effectively reduces R-loops accumulation (Figure 2(j) and (k)).
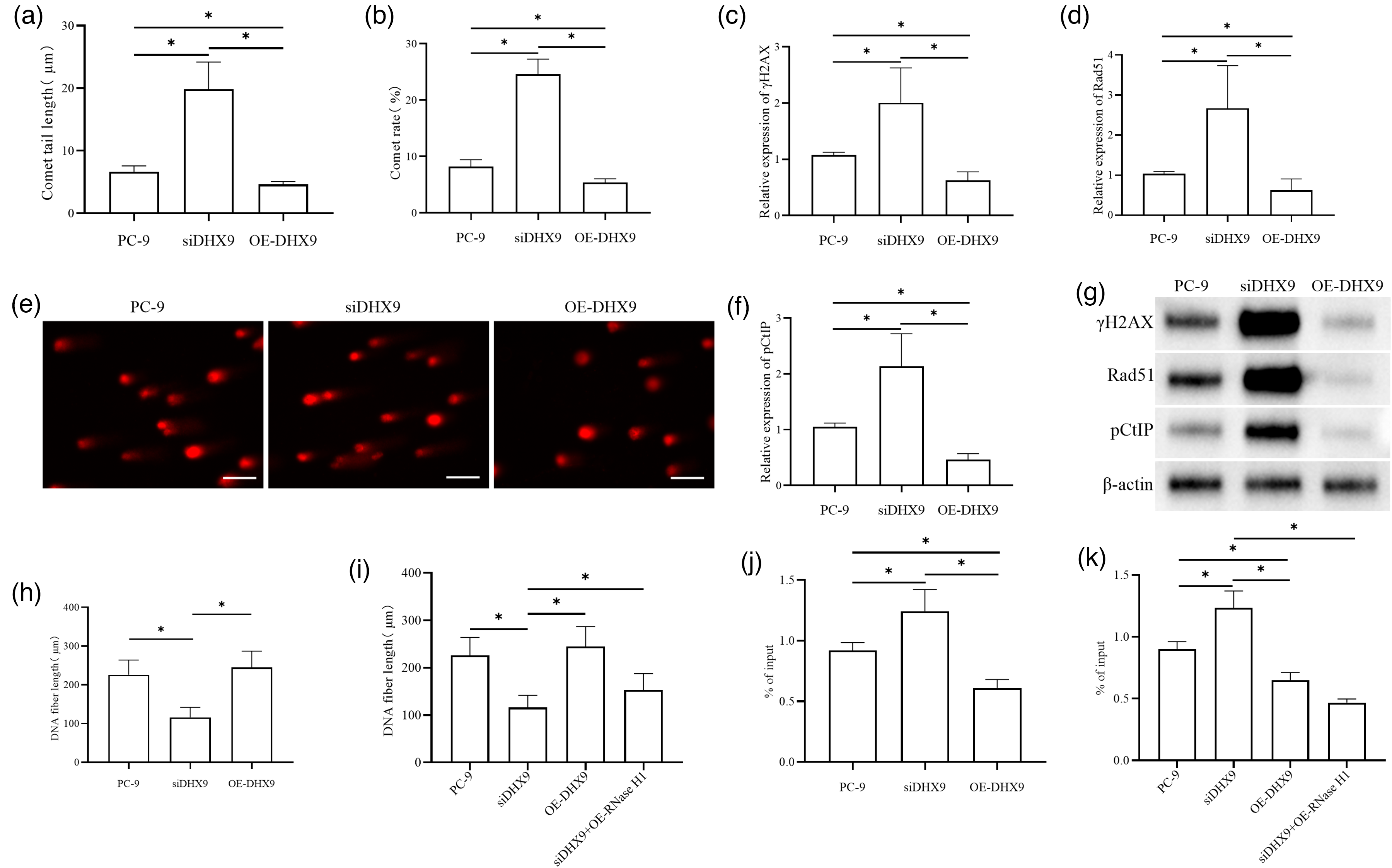
Mechanism of DHX9 in lung adenocarcinoma cells: promoting R-loop resolution and reducing DNA damage. DNA damage in lung adenocarcinoma cells after DHX9 interference was detected by comet assay (a) to (c). Western blotting was used to detect the expressions of DNA damage marker proteins and homologous recombination repair-related proteins in tumor cells after DHX9 interference (d) to (g). The representative blots are shown. The quantification of band intensity (relative to -actin) was performed using ImageJ software based on three independent experiments (d) to (f). The CldU/IdU DNA fiber pulse labeling method was applied to determine the DNA fiber lengths of lung adenocarcinoma cells in each group (h) and (i). DRIP-qPCR was used to detect R-loop accumulation in lung adenocarcinoma cells of each group (j and (k). *P < 0.05.
Phosphorylation of DHX9 at S279 inhibited R-loop resolution
To investigate whether DHX9 phosphorylation inhibits R-loop resolution, cells were transfected and divided into control, OE-PKA, DHX9-S279A, and 6-22 Amide groups. R-loop accumulation was detected by DRIP-qPCR, DNA replication status by CldU/IdU fiber labeling, and DNA damage by comet assay. DRIP-qPCR showed significantly higher DRIP enrichment in the OE-PKA group versus the PC-9 group, with lower enrichment in the DHX9-S279A group versus both PC-9 and OE-PKA groups (Figure 3(a)); the 6-22 Amide group also had significantly lower DRIP enrichment versus the PC-9 and OE-PKA groups (Figure 3(b)). CldU/IdU labeling revealed significantly shorter DNA fibers in the OE-PKA group versus the PC-9 group (no significant difference between the DHX9-S279A and PC-9 groups), while the 6-22 Amide group had significantly longer fibers versus the PC-9 and OE-PKA groups (Figure 3(c) and (d)). Comet assay demonstrated significantly longer tail length and higher comet rate in the OE-PKA group versus the PC-9 group, with lower values in the DHX9-S279A group versus both the PC-9 and OE-PKA groups (Figure 3(e) to (g)); the 6-22 Amide group also showed significantly lower tail length and comet rate versus the PC-9 and OE-PKA groups (Figure 3(h) to (j)).
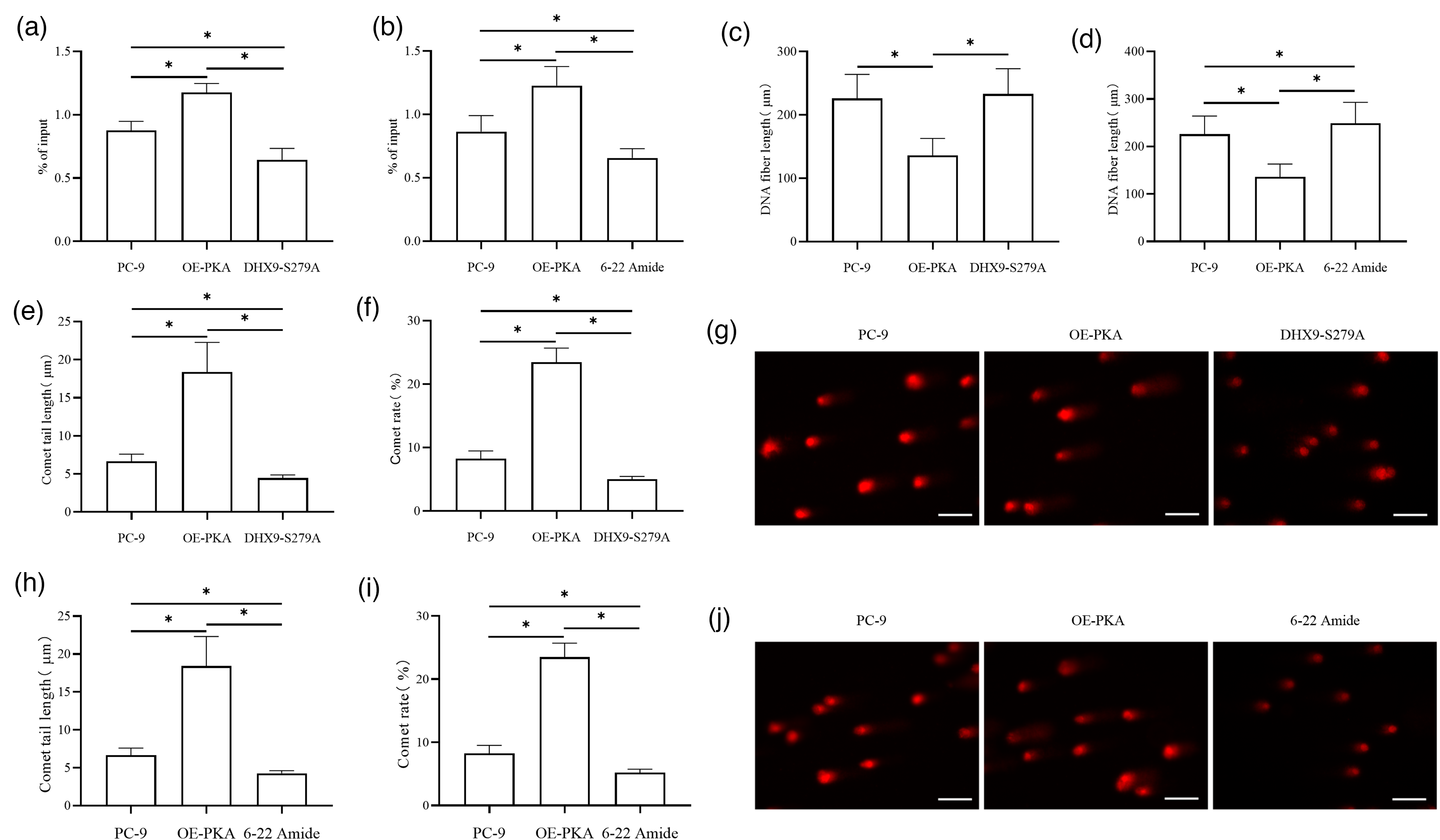
Phosphorylation of DHX9 at S279 inhibits R-loop resolution. DRIP-qPCR was used to detect R-loop accumulation in lung adenocarcinoma cells of each group (a) and (b). The CldU/IdU DNA fiber pulse labeling method was applied to determine DNA fiber lengths in each group (c) and (d). DNA damage in lung adenocarcinoma cells after DHX9 interference was detected by comet assay (e) to (g). DNA damage in lung adenocarcinoma cells after DHX9 interference was detected by comet assay (h) to (j). *P < 0.05.
Phosphorylated DHX9 at S279 inhibited lung adenocarcinoma cell proliferation through R-loop disruption
To investigate whether DHX9 phosphorylation regulates R-loop resolution to inhibit lung adenocarcinoma cell proliferation, cells were transfected and divided into control, DHX9-S279A, and DHX9-S279A + OE-RNase H1 groups, with proliferation assessed by EdU staining and CCK-8 assay, and apoptosis by TUNEL staining. EdU staining showed significantly higher proliferative activity in the DHX9-S279A group versus the PC-9 group, and even higher activity in the DHX9-S279A + OE-RNase H1 group versus both the PC-9 and DHX9-S279A groups (Figure 4(a) and (b)). CCK-8 assay revealed significantly higher OD values in the DHX9-S279A group versus the PC-9 group, with the DHX9-S279A + OE-RNase H1 group showing significantly higher proliferative activity versus both controls (Figure 4(c)). TUNEL staining demonstrated significantly lower apoptosis rates in the DHX9-S279A group versus the PC-9 group, and even lower rates in the DHX9-S279A + OE-RNase H1 group versus both the PC-9 and DHX9-S279A groups (Figure 4(d) and (e)).
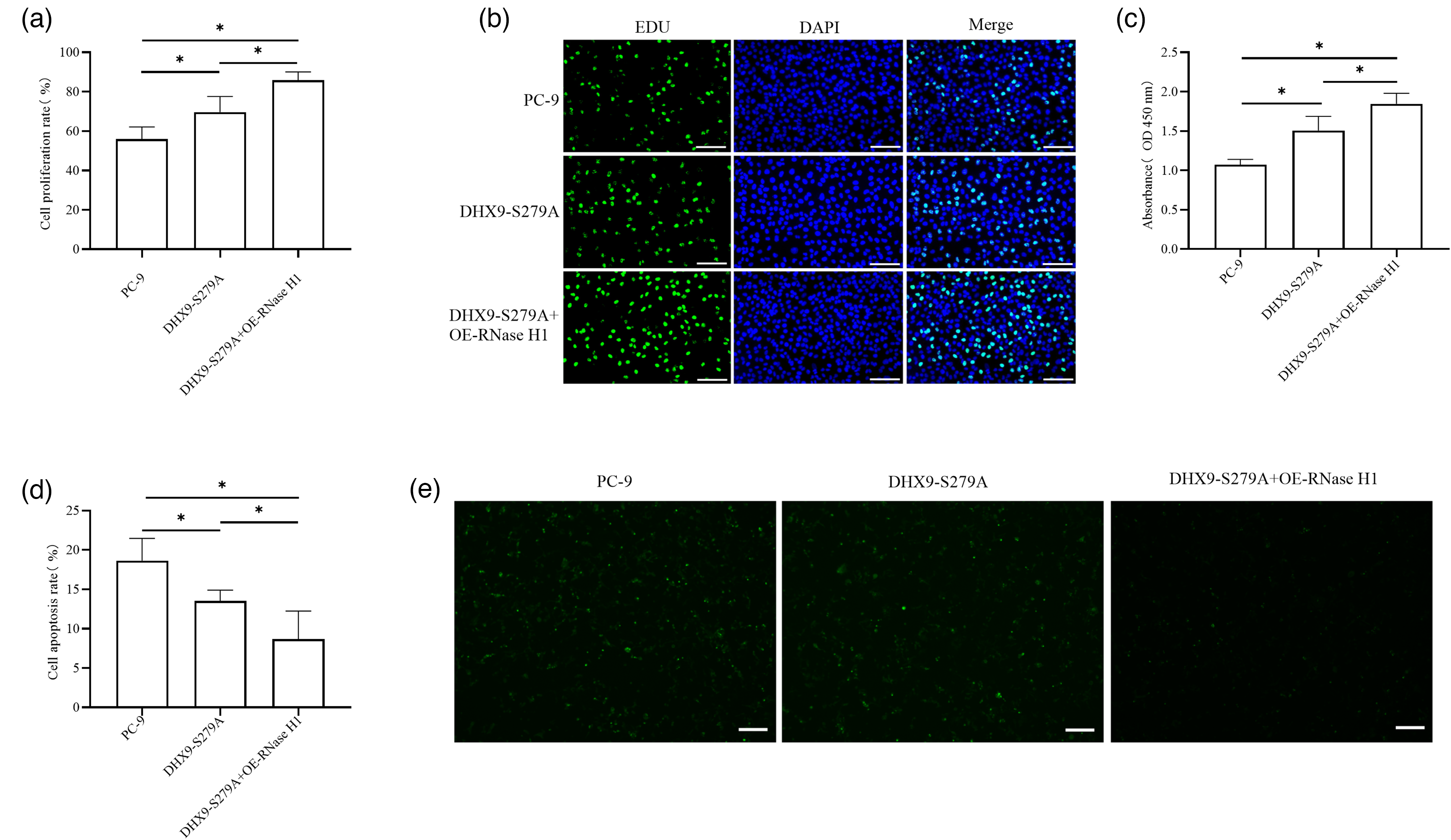
Phosphorylated DHX9 at S279 inhibits lung adenocarcinoma cell proliferation through R-loop disruption. EdU staining was used to assess the effects of various treatments on lung adenocarcinoma cell proliferation (a) and (b). The CCK-8 assay was employed to detect the impact of different treatments on lung adenocarcinoma cell proliferation (c). TUNEL staining was used to evaluate the effects of various treatments on lung adenocarcinoma cell apoptosis (d) and (e). The cell proliferation was detected by CCK-8 assay and the cell apoptosis was evaluated by TUNEL staining at 48 h after transfection. The absorbance (OD) values presented represent the data collected at 48 h after transfection. *P < 0.05.
The effect of DHX9 on R-loops and DNA damage in vivo
To verify DHX9's effects on R-loops and DNA damage, a nude mouse tumor-bearing model was established, with groups divided into control, siDHX9, DHX9-S279E, and DHX9-S279A. After 4 weeks of feeding, tumor volume and mass were measured; additional analyses included immunohistochemistry (Ki67 expression), DRIP-qPCR (R-loop accumulation), CldU/IdU labeling (DNA replication), comet assay (DNA damage), and Western blotting (γH2AX, Rad51, pCtIP). Results showed significantly smaller tumor volume and mass in the siDHX9 and DHX9-S279E groups versus the PC-9 group, with larger tumors in the DHX9-S279A group versus the siDHX9 and DHX9-S279E groups (Figure 5(a) and (b)). Immunohistochemistry revealed lower Ki67 levels in the siDHX9 and DHX9-S279E groups versus the PC-9 group, and higher levels in the DHX9-S279A group versus all other groups (Figure 5(c) and (d)). DRIP-qPCR demonstrated higher DRIP enrichment in the siDHX9 and DHX9-S279E groups versus the PC-9 group, and lower enrichment in the DHX9-S279A group versus all controls (Figure 5(e)). CldU/IdU labeling indicated shorter DNA fibers in the siDHX9 and DHX9-S279E groups versus the PC-9 group, with no significant difference between the DHX9-S279A and PC-9 groups but longer fibers versus the siDHX9 and DHX9-S279E groups (Figure 5(f)). Comet assay showed longer tail lengths and higher comet rates in the siDHX9 and DHX9-S279E groups versus the PC-9 group, and lower values in the DHX9-S279A group versus all controls (Figure 5(g) and (h)). Western blotting revealed higher γH2AX, Rad51, and pCtIP levels in the siDHX9 and DHX9-S279E groups versus the PC-9 group, with lower levels in the DHX9-S279A group versus all other groups (Figure 5(i) to (l)).
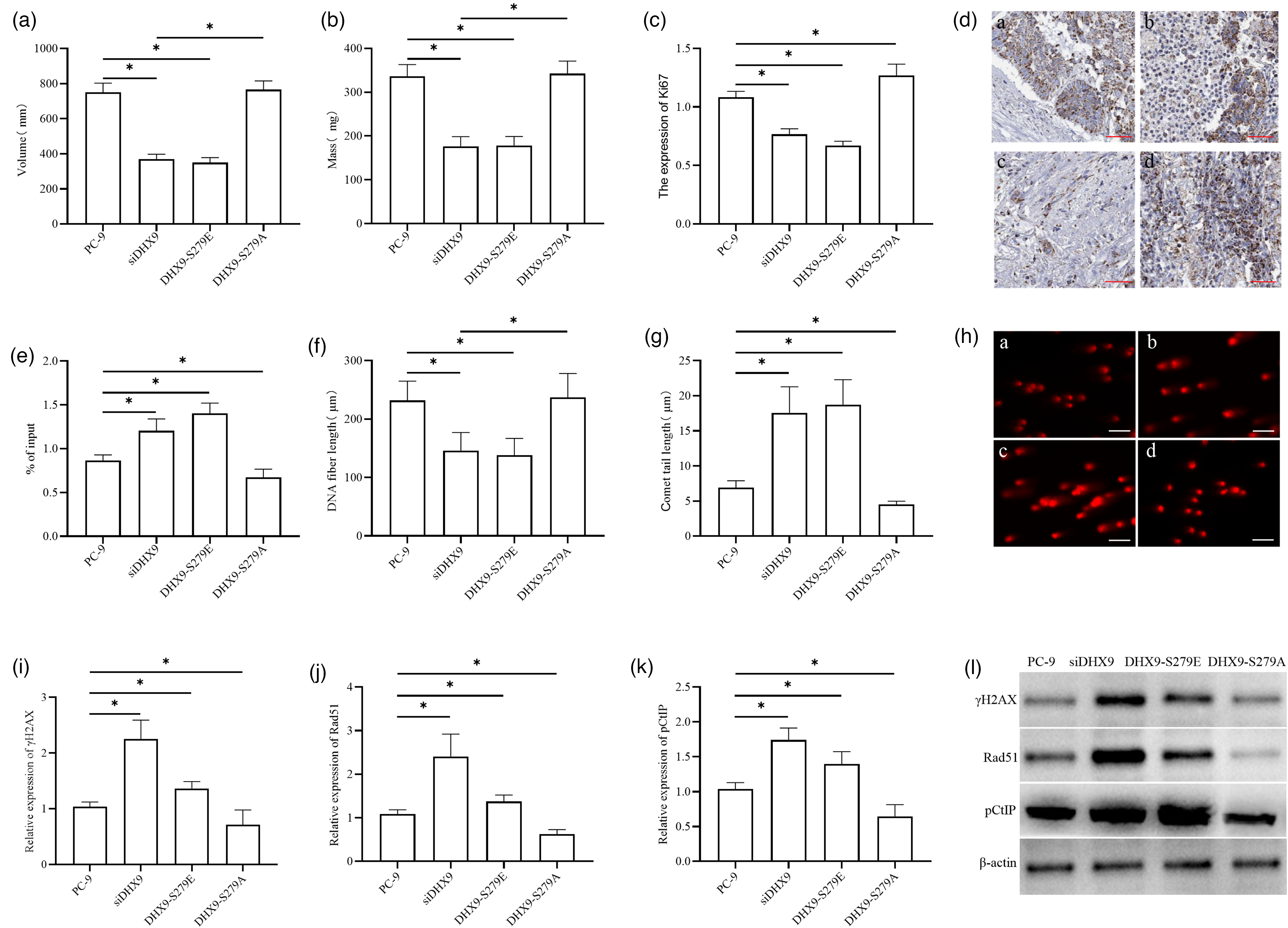
The effect of DHX9 on R-loops and DNA damage in vivo. Tumor volume (a). Tumor mass (b). Immunohistochemical detection of Ki67 expression in tumor cells (a: PC-9; b: siDHX9; c: DHX9-S279E; d: DHX9-S279A) (c) and (d). DRIP-qPCR analysis of R-loop accumulation in lung adenocarcinoma cells from each group (e). CldU/IdU DNA fiber pulse labeling for assessing DNA replication status in lung adenocarcinoma cells of each group (f). Comet assay for detecting DNA damage in lung adenocarcinoma cells of each group (g) and (h). Western blotting analysis of DNA damage marker γH2AX and homologous recombination repair-related proteins Rad51 and pCtIP expressions (i) to (l). *P < 0.05.
Discussion
Through multi-dimensional experiments, this study demonstrated that DHX9 acts as a key regulator of proliferation and apoptosis in lung adenocarcinoma cells. In qRT-PCR assays, DHX9 mRNA expression was significantly higher in the lung adenocarcinoma cell line PC-9 compared to 2BS cells. This finding was consistent with protein-level data from the Protein Atlas, thereby supporting the pro-oncogenic role of DHX9 in lung adenocarcinoma. Notably, DHX9 was increasingly recognized as a critical modulator of various cellular processes, many of which were frequently dysregulated in cancer. 14 The DHX9 protein exhibited high expression across a spectrum of malignant tumors, implying that it might act as a pan-cancer-associated molecule implicated in the initiation and progression of multiple cancer types.4,5,15 DHX9 participated in EGFR-mediated transcriptional activation. Since EGFR lacks a DNA-binding domain, DHX9 mediated this interaction through simultaneous binding to both EGFR and the promoter ATRS. 16 The expression of DHX9 in lung adenocarcinoma tissues was significantly higher than in adjacent normal tissues, and DHX9 modulated the proliferation and metastasis of cancer cells through the regulation of EMT. 4 Two independent studies analyzing DHX9 levels across panels of lung cancer samples demonstrated that DHX9 is overexpressed in tumor samples relative to normal lung tissues,17,18 with such overexpression correlating with poorer patient survival outcomes. 18 As an RNA helicase, DHX9 was likely to regulate DNA damage repair, transcriptional control, or RNA metabolism in cancer, thereby promoting tumor cell proliferation, invasion, and resistance to apoptosis.
Over the past few years, accumulating evidence indicated that R-loops were a double-edged sword. Recent studies further demonstrated that excessive R-loop accumulation contributes to genome instability and cancer progression.19,20 While R-loops functioned as critical regulatory elements in gene expression and genomic stability, their abnormal or unintended formation was associated with various diseases. Pathological R-loop formation and accumulation could act as sources of DNA damage, which are typically linked to genomic instability by exacerbating replication fork stalling and transcription-replication collisions. This was mediated through the generation of fragile single-stranded DNA (ssDNA) and double-strand breaks, as well as recombination and genomic instability arising from reduced levels or activity of topoisomerase 1. 21 Persistent R-loops could also drive heterochromatin formation and induce gene silencing. Importantly, due to the susceptibility of exposed ssDNA to DNA damage, unscheduled R-loops were increasingly recognized as a source of genomic instability—a hallmark of human cancers.8,22–24 Previous studies had reported that DHX9 maintained genomic stability by unwinding R-loops, with its depletion causing replication stress, and inhibiting proliferation. 10 In small cell lung cancer, DHX9 depletion led to abnormal R-loop accumulation, which in turn increased cytoplasmic DNA derived from DNA damage and replication stress; in vivo experiments showed that DHX9 depletion slowed tumor growth, induced a more immunogenic tumor microenvironment, and enhanced responsiveness to immune checkpoint blockade therapy. 25
Our study found that in lung adenocarcinoma PC-9 cells, DHX9 reduced DNA damage and preserved replication stability by promoting R-loops resolution. DHX9 depletion resulted in R-loop accumulation and genomic instability, with significantly elevated R-loop levels in the si-DHX9 group directly correlating with increased DNA damage and heightened replication stress. DHX9-dependent R-loop clearance was critical for sustaining replication progression, as overexpression of RNase H1 in the context of DHX9 silencing partially restored DNA replication capacity—indicating that R-loop accumulation was the primary driver of replication impairment induced by DHX9 depletion. Following DHX9 silencing, the expression levels of γH2AX, Rad51, and pCtIP proteins all increased, suggesting that persistent R-loop stress triggered the DNA damage response. Suzuki et al. 26 found that TUG1 directly and functionally interacted with DHX9 and RPA32, which could recognize ssDNA at or near R-loop regions.
Our further findings showed that the phosphorylated state of DHX9 might inhibit its R-loop unwinding activity, whereas the DHX9-S279A mutation (dephosphorylated state) or 6-22 Amide treatment enhanced R-loop clearance capacity. This, in turn, reduced DNA damage in PC-9 cells, maintained DNA replication stability, significantly improved PC-9 cell proliferation, and decreased apoptosis rates—suggesting that phosphorylation of DHX9 at the S279 site might suppress tumor progression by regulating R-loop homeostasis. Nude mouse xenograft experiments confirmed that DHX9 could influence genomic stability and participate in DNA damage repair by regulating R-loops, thereby modulating tumor growth, with the phosphorylation state of its S279 site playing a critical regulatory role in this process.
Studies revealed that DHX9 phosphorylated at the S321 site exhibited enhanced binding to γH2AX, BRCA1, and RPA, with the direct interaction with RPA being critical. Phosphorylated DHX9 strengthened its binding to RPA, thereby promoting DHX9 localization to R-loops and clearing abnormal structures via its unwinding activity, ultimately maintaining genomic stability in HeLa and U2OS cells. ATR directly regulated DHX9 through phosphorylation to eliminate stress-induced R-loops and preserve genomic stability. Expression of the non-phosphorylatable DHX9 S321A mutant prevented DHX9 interaction with RPA and R-loops, leading to the accumulation of stress-induced R-loops. 7 In colorectal cancer, oxaliplatin induced the phosphorylation of DHX9 at the S279 and S321 sites, which specifically upregulated the oncogenic circular RNA CCDC66 and ultimately enhanced tumor cell chemoresistance. 27
It is worth noting that the regulation of R-loops by DHX9 has significant clinical implications, particularly in the context of chemotherapy resistance. Recent studies have highlighted that R-loop accumulation can be induced by chemotherapeutic agents, such as Topoisomerase I inhibitors, leading to genomic instability.28,29 In colorectal cancer, oxaliplatin treatment has been shown to induce DHX9 phosphorylation, which paradoxically contributes to chemoresistance by regulating circular RNA expression. 27 Our findings regarding the S279 phosphorylation site add a new layer of complexity to this regulatory network. While the ATR-mediated phosphorylation at S321 promotes R-loop resolution to protect cells from stress, 9 our data suggest that S279 phosphorylation acts as a functional switch that suppresses DHX9's helicase activity. These findings suggest that modulation of DHX9 phosphorylation may represent a potential therapeutic avenue for sensitizing tumor cells to replication stress-inducing therapies. 30 By preventing the dephosphorylation of S279, or selectively inhibiting the kinases responsible for this modification, it may be possible to force cancer cells into a state of persistent R-loop toxicity and replication stress, thereby enhancing the efficacy of current treatments.
Our study found that phosphorylation of DHX9 at the S279 site promoted apoptosis in lung adenocarcinoma cells. We hypothesized that when the S279 site was dephosphorylated, DHX9-RPA interaction was enhanced, and DHX9 bound to R-loops, leading to R-loop removal and genomic stabilization, thereby promoting lung adenocarcinoma cell proliferation. In contrast, S279-phosphorylated DHX9 was unable to interact with RPA and R-loops, thus inhibiting lung adenocarcinoma cell proliferation. We hypothesized that phosphorylation at different sites exerted distinct effects on DHX9 function. Phosphorylation of DHX9 at S279 not only affected its intrinsic activity but it might also further regulate DNA damage responses and transcriptional processes through interactions with RPA and RNAP2. DHX9 dephosphorylation might enhance its interaction with RPA and R-loops, facilitating R-loop removal and maintaining genomic stability. Conversely, S279 phosphorylation reduced DHX9's binding capacity to RPA and R-loops, potentially impeding DNA damage repair or disrupting normal RNAP2-mediated transcription, leading to apoptosis in lung adenocarcinoma cells.
Conclusions
DHX9 is upregulated in the lung adenocarcinoma cell line PC-9, where it promotes cell proliferation and inhibits apoptosis. Additionally, DHX9 facilitates R-loop resolution and reduces DNA damage in lung adenocarcinoma cells. However, phosphorylation of DHX9 inhibits its R-loop resolution function, and phosphorylated DHX9 suppresses lung adenocarcinoma cell proliferation by disrupting R-loops. In vivo experiments using nude mice confirm the effects of DHX9 on R-loops and DNA damage, revealing that phosphorylated DHX9 may enhance DNA damage by inhibiting R-loop resolution, ultimately suppressing the proliferation of lung adenocarcinoma cells.
Supplemental Material
sj-docx-1-jbm-10.1177_03936155261433188 - Supplemental material for Phosphorylated DHX9 inhibited the progression of lung adenocarcinoma by regulating R-loops mediated DNA damage
Supplemental material, sj-docx-1-jbm-10.1177_03936155261433188 for Phosphorylated DHX9 inhibited the progression of lung adenocarcinoma by regulating R-loops mediated DNA damage by Lei Wu, MB, Shengyu Wang, MD, Xin Diao, MM, Xuan Ma, MM and Yanfeng Liu, MM in The International Journal of Biological Markers
Supplemental Material
sj-docx-2-jbm-10.1177_03936155261433188 - Supplemental material for Phosphorylated DHX9 inhibited the progression of lung adenocarcinoma by regulating R-loops mediated DNA damage
Supplemental material, sj-docx-2-jbm-10.1177_03936155261433188 for Phosphorylated DHX9 inhibited the progression of lung adenocarcinoma by regulating R-loops mediated DNA damage by Lei Wu, MB, Shengyu Wang, MD, Xin Diao, MM, Xuan Ma, MM and Yanfeng Liu, MM in The International Journal of Biological Markers
Footnotes
Acknowledgements
Not applicable.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Xi'an Municipal Bureau of Science and Technology, (grant number 23YXYJ0182).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.